
Peer Reviewed
Cleidocranial Dysplasia
Authors:
Rachel Locke, BS
Student, University of Florida, Gainesville, Florida
Kathryn Wheeler, MD
Assistant Professor of Pediatrics, University of Florida, Gainesville, Florida
Diane Howell, MD
Assistant Professor of Pediatrics, University of Florida, Gainesville, Florida
Citation:
Locke R, Wheeler K, Howell D. Cleidocranial dysplasia [published online March 18, 2019]. Consultant360.
A pregnant 34-year-old, gravida 4, para 3 woman was referred for specialty obstetric care due to abnormalities noted on a prenatal ultrasonogram at 21 weeks of gestation. The ultrasonogram had demonstrated a male fetus with an absent nasal bone, an abnormal cranial shape, shortened long bones without evidence of bowing or abnormal mineralization, and increased nuchal edema. Another ultrasonogram at 24 weeks had shown abnormal ossification of the skull, relative dolichocephaly, an enlarged posterior fossa with enlarged sutures, and minimal skull calcification.
There was no history of cranial or skeletal anomalies in the mother, the father, or their 3 offspring. Genetic testing was recommended for likely skeletal dysplasia. The woman declined amniocentesis, but the results of analysis of circulating cell-free DNA in maternal blood were normal, suggesting no chromosomal abnormalities. The pregnancy was otherwise uncomplicated, with normal fetal activity and amniotic fluid levels throughout the pregnancy.
The boy was born at 38 weeks and 4 days of gestation via scheduled cesarean delivery. The delivery was uncomplicated, the birth weight was appropriate for gestational age, and the Apgar score was 9 at 1 minute and 9 at 10 minutes. The patient was admitted to the neonatal intensive care unit (NICU) for further evaluation.
Physical examination. The neonate had frontal bossing, low-set ears, wide-spaced fontanelles with a connection between the anterior and the posterior fontanelle, small skull bones, increased nuchal thickness, lip ankyloglossia, and bone nodules on the upper maxillary dental ridge. He also had peripheral pulmonary stenosis and a webbed penis. The patient had mildly increased lower-extremity tone and mildly to moderately decreased central tone but appropriate muscle mass in all extremities. A sandal gap deformity of the feet was noted bilaterally.
Based on interpretation of the skeletal findings, his bones appeared to have stopped developing at 9 to 12 weeks of gestation. The initial suspicion was for hypophosphatasia, but the alkaline phosphatase level was normal at 70 U/L. Other considerations in the differential diagnosis included achondrogenesis, osteogenesis imperfecta, or other skeletal dysplasia.
Diagnostic tests. A radiographic skeletal survey demonstrated skeletal dysplasia with absent clavicles (Figure 1), uncalcified skull bones with an absent nasal bone (Figure 2), a bowing deformity of the clivus, sandal gap deformity (Figure 3), and an absent cisterna magna.
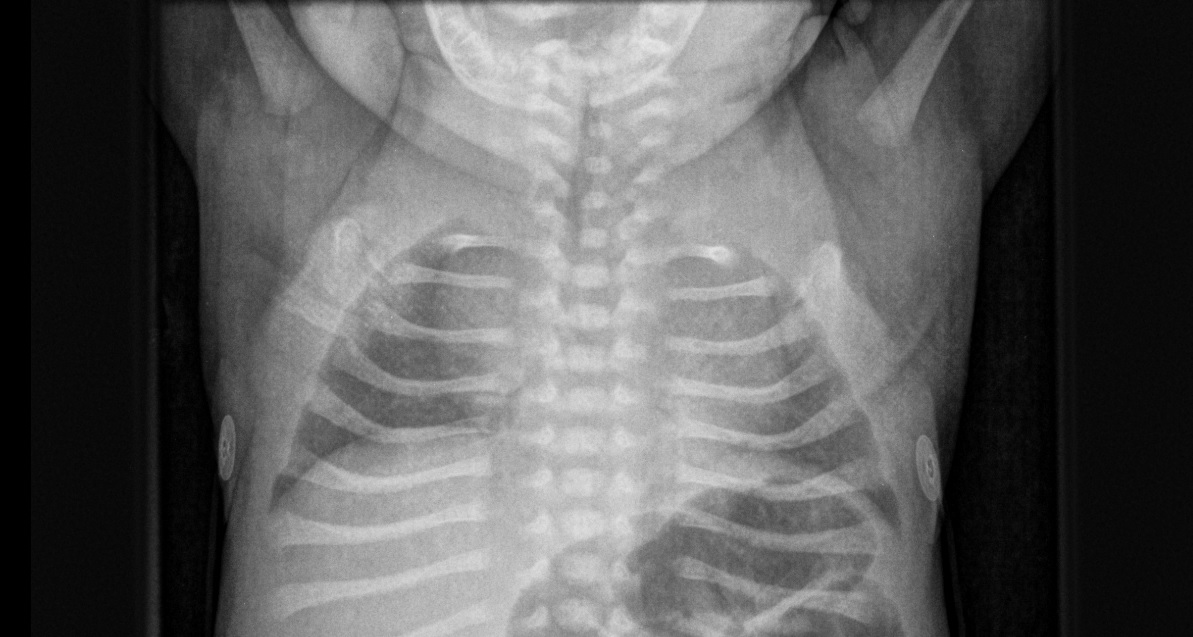
Figure 1. Chest radiograph demonstrating absent clavicles.
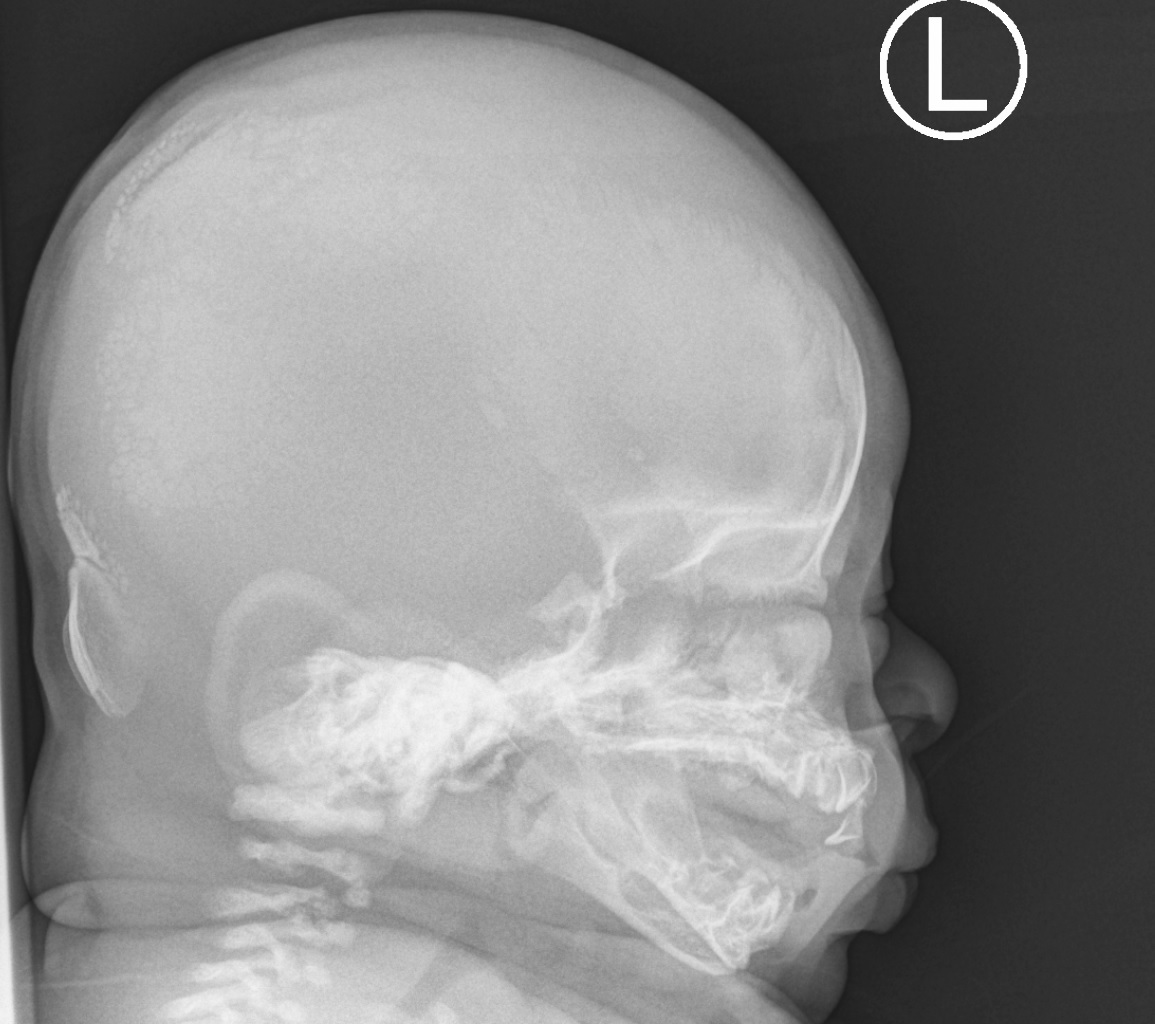
Figure 2. Skull radiograph demonstrating dolichocephaly, delayed calcification of skull bones, and absent nasal bone.
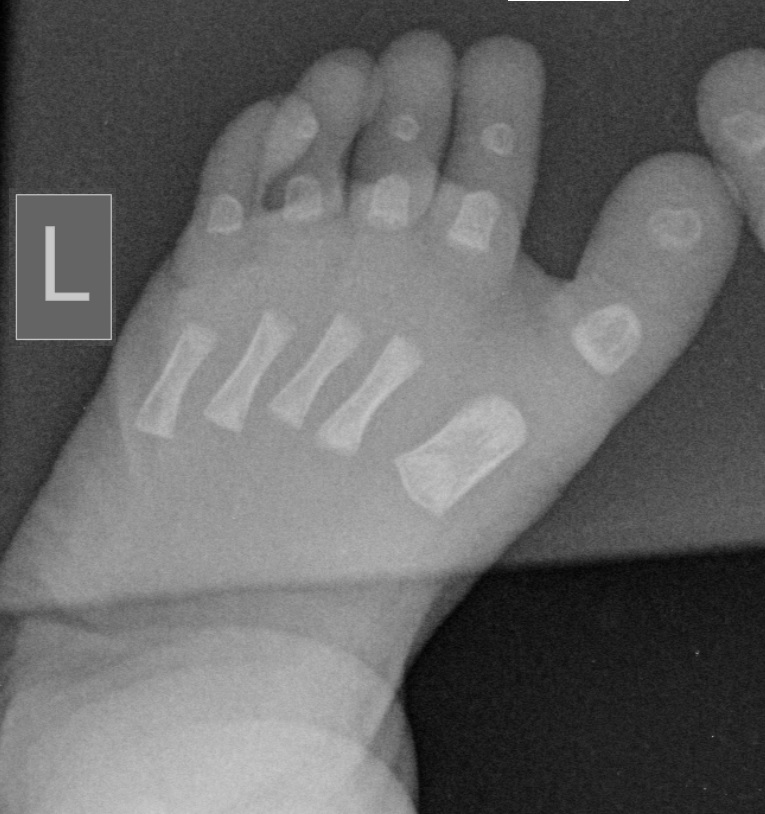
Figure 3. Radiograph of the left foot demonstrating a sandal gap deformity.
In addition to these significant findings, no fractures were present, and the vertebral bodies were calcified, making a diagnosis of osteogenesis imperfecta or achondrogenesis unlikely. The pattern of abnormalities instead suggested a diagnosis of cleidocranial dysplasia (CCD). Genetic testing for CCD was performed, including a deletion/duplication analysis of the runt-related transcription factor 2 gene (RUNX2) via array comparative genomic hybridization. The results showed a heterozygous 48.27-kb deletion including exons 7 and 9, which confirmed the diagnosis of CCD.
Discussion. Classic CCD is characterized by the triad of delayed closure of the cranial sutures, hypoplastic or aplastic clavicles, and dental abnormalities. CCD is caused by a mutation in RUNX2, which codes for a transcription factor for osteoblast development. Abnormalities can include sequence variation or a deletion or duplication within the gene. It is inherited via autosomal dominant transmission with a 50% risk for each offspring, although many cases are caused by a de novo mutation in RUNX2.1 The unusual partial deletion of one RUNX2 copy and the absence of skeletal changes in the parents suggests a de novo mutation in our patient.
Intelligence in patients with CCD is usually normal, and the most significant concern is for blunt trauma to the brain due to the lack of skull ossification. Protective helmeting is recommended during infancy and childhood. Monitoring of bone density is also important, and calcium and vitamin D supplementation should be considered. Patients should begin dual-energy X-ray absorptiometry scans in early adolescence to evaluate for osteoporosis and should receive subsequent surveillance every 5 to 10 years.1
Early referral to dental professionals is recommended, because malocclusion of the teeth and jaws is common. Patients with CCD often have supernumerary teeth due to delayed loss of the primary teeth and delayed appearance of the secondary teeth.2 Patients can also present with cysts in the gums.
Patients with CCD are at increased risk of recurrent respiratory and ear infections.3 Respiratory infections can be complicated by the absence of the nasal bone resulting in narrow nasal cavities prone to upper airway obstruction.4 Infections should be treated aggressively, and referral to an otolaryngologist is highly recommended.
Outcome of the case. Since discharge from the NICU, our patient has been monitored closely. While his skull is unlikely to completely ossify, he has had increasing calcification over the first year of life. It is likely that he will always have one or more open fontanelles. His case is being followed by an orthotist, and he has been provided a helmet to wear when he is active or in a vehicle. Calcium and vitamin D levels have remained normal. The premaxillary cyst present at birth resolved, and his first primary teeth erupted on a typical schedule.
He underwent myringotomy and tube placement without incident at 11 months of age after his second episode of acute otitis media, which had required treatment with ceftriaxone. Results of a sleep study at 4 months of age demonstrated moderate obstructive sleep apnea unrelated to the size of the tonsils or adenoids. Additionally, he had a prolonged hospital stay in the pediatric intensive care unit at 5 months of age for concurrent respiratory syncytial virus and parainfluenza infections requiring bilevel positive airway pressure therapy.
He otherwise has had normal growth and development, but his case is being followed in an early intervention services program for high-risk surveillance.
- Machol K, Mendoza-Londono R, Lee B. Cleidocranial dysplasia spectrum disorder. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2017. http://www.ncbi.nlm.nih.gov/books/NBK1513/. Updated November 16, 2017. Accessed March 18, 2019.
- Paul SA, Simon SS, Karthik AK, Chacko RK, Savitha S. A review of clinical and radiological features of cleidocranial dysplasia with a report of two cases and a dental treatment protocol. J Pharm Bioallied Sci. 2015;7(suppl 2):S428-S43
- Cleidocranial dysplasia. Genetics Home Reference, US National Library of Medicine, National Institutes of Health. http://ghr.nlm.nih.gov/condition/cleidocranial-dysplasia. Reviewed August 2017. Accessed March 18, 2019.
- Farrow E, Nicot R, Wiss A, Laborde A, Ferri J. Cleidocranial dysplasia: a review of clinical, radiological, genetic implications and a guidelines proposal. J Craniofac Surg. 2018;29(2):382-389.