
Apert Syndrome
Photoclinic

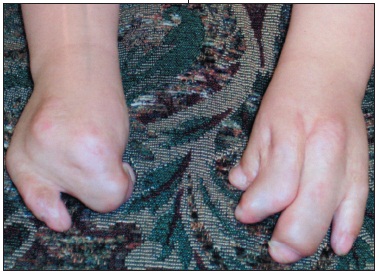
This 5-year-old girl with multiple congenital abnormalities presented for a routine checkup. She was born with Apert syndrome, also known as acrocephalosyndactyly.A peaked head, sloping eyes, and "mitten hands" are typical features.
This disorder is characterized by premature closure of cranial sutures (craniosynostosis), which results in a distorted head shape and an unusual facial appearance. The vast majority of cases are sporadic and occur spontaneously without a family history. A mutation in the fibroblast growth factor receptor 2 (FGFR2) gene, which maps to chromosome locus 10q25-10q26, causes the bony sutures of the skull to close prematurely. There may be severe webbing or fusion of several adjacent fingers and toes. As the child grows, the bones in the hands and feet become progressively fused, which reduces flexibility and function.
Children with Apert syndrome may have mental deficiencies, short stature, hearing impairment, frequent ear infections, prominent and/or bulging eyes, a large or late-closing soft spot on the skull,dental anomalies, and other skeletal and congenital abnormalities.
Other syndromes associated with craniosynostosis that have similar extracranial manifestations include:
• Crouzon syndrome (craniofacial dysostosis).
• Carpenter syndrome (kleeblattschädel syndrome, cloverleaf skull deformity).
• Saethre-Chotzen syndrome.
• Pfeiffer syndrome.
 Bhagwan Das Bang, MD, of Opp, Ala, notes that most cases are associated with older paternal age. A radiograph of the skull and clinical examination can confirm the diagnosis of craniosynostosis. Radiographs of the hands and feet can help determine the extent of bony problems. A genetic test for mutations in the FGFR2 gene confirms the diagnosis of Apert syndrome. Hearing tests should also be performed.
Bhagwan Das Bang, MD, of Opp, Ala, notes that most cases are associated with older paternal age. A radiograph of the skull and clinical examination can confirm the diagnosis of craniosynostosis. Radiographs of the hands and feet can help determine the extent of bony problems. A genetic test for mutations in the FGFR2 gene confirms the diagnosis of Apert syndrome. Hearing tests should also be performed.
Treatment consists of early surgical correction of the craniosynostosis, after evaluation by a multispecialty craniofacial surgery team at a children's medical center. An otolaryngologist can help optimize treatment of hearing problems. Support groups, such as Apert Syndrome Pen Pals, are available.
Other congenital abnormalities may coexist and patients should be evaluated on an individual basis. There is a slight risk of recurrence in unaffected parents of a child with Apert syndrome; in affected parents, the recurrence risk is 50%.
(Photographs courtesy of Renee Lemaire, marketing director, Mizell Memorial Hospital, Opp, Ala.)
FOR MORE INFORMATION:
Jones KL. Apert syndrome. In: Jones KL, ed. Smith's Recognizable Patterns of Human Malformations. 5th ed. Philadelphia: WB Saunders Co; 1997:418-419.