
Peer Reviewed
Mimicking CNS Malignancy: A Rare Case of Autoimmune GFAP Astrocytopathy
Introduction. A 72-year-old man was referred to the emergency department by his primary care physician (PCP) for evaluation of dizziness and gait instability.
History. The patient reported a 3 to 4-day history of dizziness and gait unsteadiness exacerbated by head movement to his PCP. His medical history included chronic obstructive pulmonary disease (Trelegy Ellipta), coronary artery disease status post-angioplasty (Aspirin, 81 mg), type 2 diabetes mellitus, hypertension (Metoprolol, 25 mg), dyslipidemia (Atorvastatin, 20 mg), a benign pancreatic cyst, and prostate cancer treated with prostatectomy. He denied recent infection, trauma, or constitutional symptoms.
Two weeks after his initial presentation, the patient presented again to his PCP with worsening symptoms, including new-onset diplopia and right foot weakness, prompting a head computed tomography (CT), which revealed an acute-to-subacute infarct in the right occipital and posterior parietal lobes. Magnetic resonance imaging (MRI) performed prior to admission confirmed a necrotic enhancing lesion in the right occipital lobe with surrounding edema, which was concerning for a malignancy such as primary CNS lymphoma. He was admitted for further diagnostic evaluation, including neurology and neurosurgical consultations. On examination, mild gait ataxia, a positive Romberg sign, right foot weakness, and diplopia were noted.
Diagnostic Testing. One day after admission, an initial brain MRI revealed a necrotic, contrast-enhancing lesion in the right occipital lobe with surrounding vasogenic edema, along with smaller enhancing nodules near the right lateral ventricle and in the left cerebral hemisphere (Figure 1A). Four days following admission, cervical and thoracic spine MRI demonstrated punctate enhancing foci within the proximal spinal cord and pons (Figure 1B), making findings initially suspicious for metastatic disease.
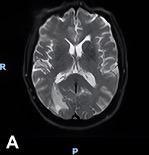
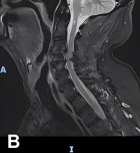
Figure 1A-B. (A) Initial brain MRI shows a necrotic, enhancing lesion with surrounding edema in the right occipital lobe. Several small enhancing nodules are seen near the right occipital and temporal horns in the left hemisphere. (B) Sagittal view of thoracic and cervical MRI with prominent focus at the level of T4 shows punctate enhancing foci scattered within the proximal spinal cord and pons, findings suspicious for metastatic disease.
Following the initial brain MRI, a lumbar puncture (LP) performed during hospitalization revealed atypical lymphocytes but no immunophenotypic evidence of lymphoma. A subsequent LP, performed 2 days later, showed persistent pleocytosis without malignant cells. Six days after his initial brain MRI, a follow-up MRI demonstrated extension of the right occipital lesion with leptomeningeal enhancement along ventricular surfaces, brainstem, and cranial nerves (Figure 2A-B), raising continued concern for lymphoma or metastatic disease.
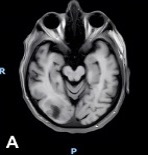
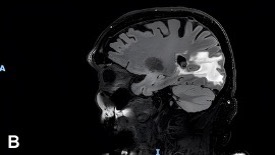
Figure 2A-B. MRI of the brain shows a right occipital lobe lesion extending to the lateral ventricle with abnormal contrast enhancement along ventricular, ependymal, and leptomeningeal surfaces. (A) Axial contrast-enhanced T1-weighted MRI demonstrating an irregular enhancing lesion that is centered in the right occipital lobe, extending toward the occipital horn of the right lateral ventricle. (B) Sagittal T2-weighted FLAIR MRI demonstrating extensive hyperintense signal within the right occipital and posterior parietal white matter that is consistent with vasogenic edema surrounding the lesion with mild local mass effect.
A right occipital craniotomy and biopsy of 2 enhancing lesions was performed 2 days after the follow-up brain MRI. A frozen section suggested lymphoma; however, final pathology demonstrated a primary demyelinating process. Cerebrospinal fluid (CSF) flow cytometry remained negative for malignancy. A presumptive diagnosis of tumefactive multiple sclerosis (MS) was considered but later dismissed due to atypical features.
Following biopsy results demonstrating a demyelinating process, high-dose intravenous dexamethasone was initiated and produced marked radiographic improvement (Figure 3), but new enhancing foci developed 1 month after corticosteroid therapy. Recurrent lesions and new-onset optic neuritis prompted additional workup. GFAP-IgG antibodies were ultimately identified in cerebrospinal fluid, confirming an autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy diagnosis.
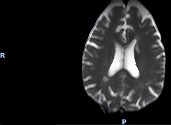
Figure 3. Post-treatment brain MRI demonstrates significant improvement and near-resolution of the right dominant occipital lesion following high-dose corticosteroid therapy.
Differential Diagnosis. The initial differential included primary CNS lymphoma, metastatic disease, and tumefactive demyelinating lesions. Malignancy was suspected due to enhancing masses and atypical lymphocytes on CSF analysis; however, malignancy became unlikely after nondiagnostic pathology and negative serial CSF cytology.
Multiple sclerosis was considered but dismissed due to the patient’s age of onset and atypical imaging features, including a brain MRI that demonstrated extensive leptomeningeal and ependymal enhancement, multifocal areas of contrast enhancement involving the ventricular margins, brainstem, and basal ganglia, as well as involvement of multiple cranial nerve branches. This pattern of diffuse leptomeningeal and ventricular surface involvement is not typical of MS and raised concern for alternative etiologies, including primary CNS lymphoma or metastatic disease.
Infectious etiologies were excluded by negative microbial studies. Ultimately, the detection of GFAP-IgG in the CSF established the diagnosis.
Treatment and Management. Upon confirmation of GFAP astrocytopathy, the patient began long-term immunosuppression with oral prednisone (70 mg daily) and mycophenolate mofetil (1 g twice daily), along with trimethoprim–sulfamethoxazole prophylaxis. He participated in an inpatient rehabilitation program that included physical, occupational, and speech therapy to address paraparesis, cognitive slowing, and gait instability.
Outcome and Follow-Up. Following combined corticosteroid and immunosuppressive therapy, the patient demonstrated gradual clinical and functional improvement. With resolution of encephalopathy and improved strength and coordination, he regained partial independence. He tolerated therapy well, with no reported adverse effects or further relapses during follow-up. Ongoing outpatient monitoring with serial MRI and neurologic evaluation was planned to assess disease stability and medication adherence.
Discussion. GFAP astrocytopathy is a newly recognized autoimmune inflammatory disorder of the CNS, first detailed by Fang et al in 2016.1 It is characterized by GFAP-specific IgG antibodies that target an astrocytic intermediate filament protein and are best detected in cerebrospinal fluid.2
The pathogenesis has been suspected to be primarily T-cell–mediated, involving GFAP-specific cytotoxic lymphocytes and abnormal tumor necrosis factor signaling.3 Cerebrospinal fluid typically reveals lymphocytic pleocytosis and elevated protein,3 consistent with the inflammatory pattern evident in our case.
The disease often presents acutely or subacutely with features of meningoencephalomyelitis, including fever, encephalopathy, ataxia, myelitis, and visual disturbances.4 Neuroimaging commonly shows periventricular or basal ganglia T2/FLAIR hyperintensities and gadolinium enhancement; however, these features overlap significantly with other CNS pathologies, including neoplastic, infectious, and demyelinating conditions. The characteristic “radial perivascular enhancement” pattern, described in the literature as suggestive of GFAP astrocytopathy, was not present in this case, highlighting diagnostic complexity and variability.5
GFAP astrocytopathy can coexist with other autoimmune conditions and neuronal antibodies (such as aquaporin-4 or NMDAR) and has been linked to paraneoplastic syndromes, particularly teratomas or carcinomas.6 Despite the patient’s history of prostate cancer, extensive malignancy workup was ultimately negative, supporting a non-paraneoplastic autoimmune mechanism. Malignancy workup included MRI of the brain and spine demonstrating multifocal enhancing lesions with leptomeningeal involvement, which initially raised concern for primary CNS lymphoma or metastatic disease. CSF analysis showed lymphocytic pleocytosis but was negative for malignant cells on cytology and flow cytometry on multiple lumbar punctures. A right occipital craniotomy with biopsy of two lesions was performed with the frozen section suggesting lymphoma, but final pathology demonstrated a primary demyelinating process without evidence of malignancy.
Treatment typically includes high-dose corticosteroids for acute management and steroid-sparing immunosuppressants such as mycophenolate, azathioprine, or rituximab for maintenance. Most reported cohorts demonstrate favorable response to corticosteroid-based therapy.6 In this case, sustained immunotherapy led to marked recovery, emphasizing the importance of early recognition and treatment initiation.
This case highlights the ability of autoimmune GFAP astrocytopathy to mimic CNS malignancy on neuroimaging. Indeed, its clinical and radiologic resemblance with other CNS disorders can lead to misdiagnosis, unnecessary surgical intervention, and ultimately poor patient outcomes. Greater clinical awareness of this disorder and appropriate antibody testing are essential for timely diagnosis and intervention.
Conclusion. Autoimmune GFAP astrocytopathy is a rare yet treatable autoimmune disorder of the CNS. Because it can mimic malignancy or demyelination, clinicians should consider it in patients with recurrent, steroid-responsive CNS lesions of unclear origin. Early detection of GFAP-IgG and prompt immunotherapy can lead to substantial recovery and prevent long-term neurological disability.
AUTHORS:
Madelyn Rayos, BS1 • Janet Park, BS, MS1 • Sashi Makam, MD2,3
AFFILIATIONS:
1Touro College of Osteopathic Medicine, Middletown, NY
2Mid Hudson Medical Research, New Windsor, New York
3Horizon Family Medical Group, New Windsor, New York
CITATION:
Rayos M, Park J, Makam S. Mimicking CNS malignancy: A rare case of autoimmune GFAP astrocytopathy. Consultant. Published online March 17, 2026. doi: 10.25270/con.2026.03.0000002
Received November 2, 2025. Accepted January 19, 2026.
DISCLOSURES:
The authors report no relevant financial relationships.
ACKNOWLEDGEMENTS:
None.
CORRESPONDENCE:
Madelyn Rayos, BS, Touro College of Osteopathic Medicine, 60 Prospect Avenue, Middletown, NY 10940. (email: mrayos@student.touro.edu)
References
- Fang B, McKeon A, Hinson SR, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. 2016;73(11):1297-1307. doi:10.1001/jamaneurol.2016.2549
- Li X, Li J, Xu H, et al. Review of clinical and imaging findings in autoimmune glial fibrillary acidic protein astrocytopathy to aid in early diagnosis. Front Immunol. 2024;15:1466847. doi:10.3389/fimmu.2024.1466847.
- Yuan Z, Li H, Huang L, et al. CD8+ T-cell predominance in autoimmune glial fibrillary acidic protein astrocytopathy. Eur J Neurol. 2021 Jun;28(6):2121-2125. doi: 10.1111/ene.14778. Epub 2021 Mar 6. PMID: 33590610.
- Kimura A, Takekoshi A, Yoshikura N, Hayashi Y, Shimohata T. Clinical characteristics of autoimmune GFAP astrocytopathy. J Neuroimmunol. 2019;332:91-98. doi:10.1016/j.jneuroim.2019.04.004.
- Kunchok A, Zekeridou A, McKeon A. Autoimmune glial fibrillary acidic protein astrocytopathy. Curr Opin Neurol. 2019;32(3):452-458. doi:10.1097/WCO.0000000000000676.
- Wei WX, Chen ML, Meng L. Case report: Autoimmune glial fibrillary acidic protein astrocytopathy with overlapping autoimmune syndrome. Front Immunol. 2024;15:1485374. doi: 10.3389/fimmu.2024.1485374.
©2026 HMP Global. All Rights Reserved.
Any views and opinions expressed are those of the author(s) and/or participants and do not necessarily reflect the views, policy, or position of Consultant360 or HMP Global, their employees, and affiliates.