
Peer Reviewed
Choledochal Cyst With Rupture and Pseudocyst Formation
AFFILIATIONS:
1Resident Physician, Pediatrics Department, Good Samaritan Hospital Medical Center, West Islip, New York
2Pediatrics Department, Good Samaritan Hospital Medical Center, West Islip New York
3Chief, Pediatric Surgery Department, Good Samaritan Hospital Medical Center, West Islip, New York
CITATION:
Ichoa R, Savargaonkar R, Jones V. Choledochal cyst with rupture and pseudocyst formation. Consultant. 2022;62(10):e21-e24. doi:10.25270/con.2022.07.00003
Received January 26, 2021. Accepted April 20, 2021. Published online July 6, 2022.
DISCLOSURES:
The authors report no relevant financial relationships. The authors report that informed patient consent was obtained for publication of the images used herein.
CORRESPONDENCE:
Reem Ichoa, DO, Pediatrics Department, BayCare Medical Group, 12780 Race Track Rd Suite 205, Tampa, FL 33626 (reemichoa@gmail.com)
ABSTRACT
Choledochal cysts are congenital anomalies of the biliary duct. The most common site for the development of cystic dilation is the common bile duct. Although choledochal cysts are a rare disease in children, complications such as perforation of the cyst have been reported. In this literature review, we discuss a case of a choledochal cyst with rupture and pseudocyst formation. Complete excision of the cyst and pseudocyst yields an excellent outcome with a 90% 5-year survival rate postoperatively.
Key words: abdominal pain, choledochal cyst, pseudocyst, emergency intervention, pediatrics
Introduction
The incidence of choledochal cyst is 1 in 100,000 to 150,000 in the Western population, making it a rare disease in young children.2 It presents most commonly in the Asian population with a female predominance.2 Spontaneous rupture and pseudocyst formation are uncommon complications. With the development in imaging modalities, it has been easier to accurately diagnose a choledochal cyst; however, it is difficult to diagnose a perforation preoperatively. Roux-en-Y hepaticojejunostomy bilioenteric reconstruction was done in this case review and it has been determined that complete resection of the cyst yields the best outcome and prognosis.
Case Presentation
A 3-year-old boy presented with abdominal pain and vomiting for 4 days. During that time, he had five or six episodes of vomiting accompanied by diffuse abdominal pain. His mother reported decreased oral intake but denied any fever, cough, or diarrhea. His symptoms continued to worsen, so he was taken to the emergency department (ED) for further evaluation. In the ED, the patient was afebrile and had stable vital signs.
Physical examination. The examination was remarkable for tenderness in the epigastric and right upper quadrant regions of the abdomen, with a palpable mass in the right upper quadrant. He had no distention or guarding. His bowel sounds were present in all four quadrants. His laboratory values are shown in Table 1.
|
Table 1. Laboratory Values of the Patient |
||
|
|
Reference Values |
|
|
White blood cell count |
15.91 × 103/mL3 |
5-15.5 × 103/mL3 |
|
Hemoglobin |
13.7 g/dL |
11.5-13.5 g/dL |
|
Hematocrit |
39.7% |
34%-40% |
|
Platelets |
454 × 103/mL3 |
150-350 × 103/mL3 |
|
Aspartate transferase |
71 IU/L |
15-40 IU/L |
|
Alanine transaminase |
82 IU/L |
5-45 IU/L |
|
Bilirubin |
0.9 mg/dL |
< 1.5 mg/dL |
|
Alkaline phosphatase |
296 IU/L |
100-320 IU/L |
|
Sodium |
134 mmol/L |
135-147 mmol/L |
|
Chloride |
6 mmol/L |
97-107 mmol/L |
|
Bicarbonate |
18 mmol/L |
22-26 mmol/L |
|
Lipase |
266 U/L |
3-32 U/L |
Imaging. An abdominal radiography scan was performed and showed unremarkable findings. A computed tomography scan of the abdomen and pelvis revealed a large cyst measuring 6.4 cm × 6.1 cm in the porta hepatic region, inseparable from the liver, pancreatic head, gallbladder, and duodenum (Figures 1 and 2), suggestive of a choledochal cyst. No bowel obstruction was identified.
Treatment. The patient was treated with an antiemetic and intravenous fluid hydration. He was then admitted to the pediatric department for further management of his choledochal cyst. Magnetic resonance imaging without contrast was performed upon admission and showed a large cystic lesion in the porta hepatis communicating with the gallbladder, consistent with a choledochal cyst type IA. No intrahepatic ductal dilatation was identified. A small nonobstructing calculus was noted in the gallbladder as well as some debris within the cystic lesion. A small amount of pericholecystic and right lower quadrant free fluid was also present.
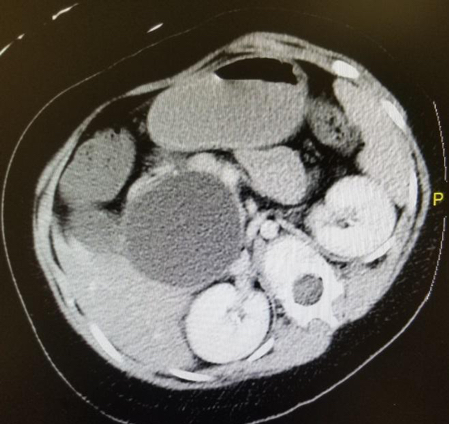
Figure 1. CT of the patient’s abdomen and pelvis revealing choledochal cyst (cross-section).
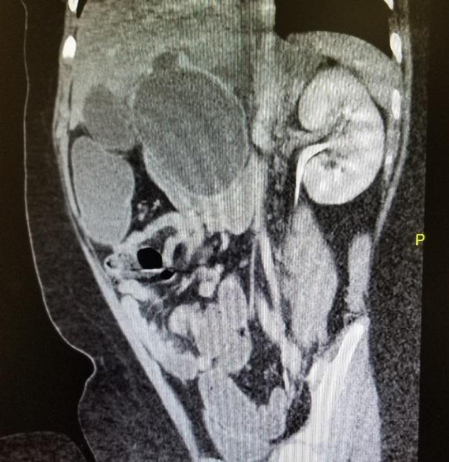
Figure 2. CT of the patient’s abdomen and pelvis revealing choledochal cyst (coronal section).
Magnetic resonance cholangiopancreatography (MRCP) was performed to delineate the cyst. Choledochal cyst resection with Roux-en-Y hepaticojejunostomy was performed on day 4 of hospitalization. During the surgery, the gallbladder was noted to be dilated. The cystic duct was also dilated and tortuous, extending up to the common hepatic duct. The cyst had evidence of contained perforation into the lesser sac with formation of a pseudocyst measuring 10 cm in diameter (Figure 3).
Patient outcome. The patient tolerated the surgery well. A nasogastric (NG) tube was placed and peripheral parenteral nutrition was started. Antibiotics were continued during his hospital stay, and his pain was well controlled. Follow-up liver enzymes and lipase levels significantly improved. On postoperative day 5, the NG tube was removed, and the patient could tolerate a liquid diet. His diet was then advanced, and he was stable for discharge on postoperative day 8.
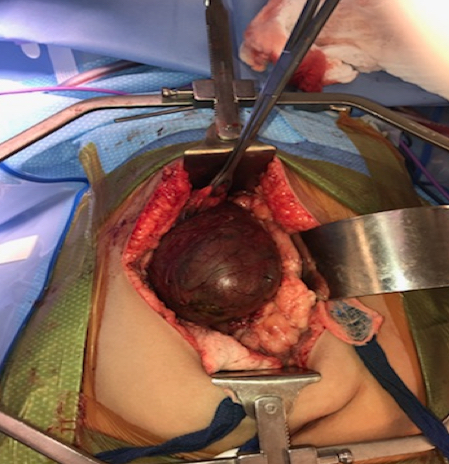
Figure 3. The choledochal cyst intraoperatively.
Discussion
Choledochal cysts are rare, with an incidence of 1 per 100-150,000 in the Western population.1 It is primarily seen in the Asian population, with an incidence of 1 per 1000.2 It remains relatively uncommon in the United States, affecting 1 in 13,500 live births.1 The female-to-male ratio ranges from 3:1 to 4:1.2 About 80% of cases are diagnosed before the age of 10 years; if not treated, serious complications can develop.1
According to the Todani classification, choledochal cysts are classified into five types (Table 2).1 Type I is the most common type, followed by type IV A.1,6 The exact etiology of choledochal cyst is unknown; however, the most accepted theory is the formation of the long common channel secondary to a pancreaticobiliary junction abnormality.1 A long common channel is defined by some authors as greater than 15 mm from the insertion of the common bile duct to the ampulla of Vater. If the junction is at any level above the duodenal wall, the common channel is considered long. It is because any length will allow for the mixing of the pancreatic secretions and bile leading to reflux and inflammation of the wall of the biliary tract. Dilation then develops due to the buildup of the pressure in the channel, leading to a weak cystic wall.1
|
Table 2. Todani Classification of Choledochal Cysts |
||||
|
I |
IA |
Extrahepatic cystic biliary dilation |
|
|
|
IB |
Extrahepatic saccular biliary dilation |
|||
|
IC |
Extrahepatic fusiform biliary dilation |
|||
|
II |
Extrahepatic biliary diverticulum |
|
||
|
III |
Choledochocele: intraduodental diverticulum |
|
||
|
IV |
IVA |
Multiple cystic dilation of the extrahepatic and intrahepatic bile duct |
|
|
|
IVB |
Multiple cystic dilation of only the extrahepatic bile duct |
|
||
|
V |
Multiple cystic dilation of only the intrahepatic bile duct |
|
||
|
Note: This table has been recreated from data taken from Altıntoprak F, Uzunoğlu MY, Dikicier E, Zengin I. Choledochal cysts—Classification, physiopathology, and clinical course. Integr Cancer Sci Therap. 2016;3(5):588-592. doi:10.15761/ICST.1000209 |
||||






Complications of choledochal cysts include acute cholangitis, perforation, pancreatitis, hepatolithiasis, and malignancy of the biliary tract. Biliary pseudocysts can form if perforation of the choledochal cysts occur. This is considered a very rare complication, especially in the pediatric population. The incidence of a spontaneous perforation of choledochal cysts is 1.8% to 7%.2 There are only a few cases of biliary pseudocysts formation reported in literature, including a case in an infant with very low birth weight and another diagnosed in a newborn with rupture occurring prenatally.2,3
Pseudocyst formation following choledochal cyst perforation typically presents with abdominal pain and jaundice, which frequently cannot be distinguished from a nonperforated choledochal cyst.4 It is also difficult to diagnose perforation preoperatively. For these reasons, perforation of the choledochal cyst cannot be excluded based on the clinical presentation and imaging obtained. Another serious complication following perforation of biliary tree is biliary peritonitis.5 Although it is rare and more commonly seen with duodenal ulcer perforation, it is an intra-abdominal emergency and requires immediate intervention. Choledochal cyst malignancy is rare in the pediatric population and is commonly associated with types I and IV cysts. The most common associated malignancies are adenocarcinomas, anaplastic carcinomas, undifferentiated carcinomas, and squamous cell carcinomas.1 The extrahepatic biliary tract is the most common site for these malignancies, followed by the gallbladder, liver, and pancreas.1 Choledochal cysts are also associated with a variety of congenital anomalies, such as duodenal atresia, colonic atresia, imperforate anus, pancreatic arteriovenous malformation, multiseptate gallbladder, ventricular septal defect, and pancreatic aplasia.1
With the development seen in the imaging modalities, choledochal cysts are often diagnosed incidentally. Ultrasound is noninvasive, reliable, and easy to use. Its low cost makes it the most frequently used imaging modality in the pediatric population. MRCP is another noninvasive modality with a sensitivity of 70% to 100% and specificity of 90% to 100%.6 It is currently the imaging study of choice to diagnose and classify choledochal cysts.6 It delineates the anatomy accurately and helps plan the extent of the operative resection. MRCP has replaced cholangiography as a diagnostic test for choledochal cysts but cannot be used for therapeutic purposes.6
Differential diagnosis. There were a variety of differential diagnoses to consider regarding the right upper quadrant mass prior to performing the MRCP. Differential diagnoses included neuroblastoma, hepatoblastoma, small bowel obstruction, hydronephrosis, and polycystic kidney disease.
Appropriate therapy of choledochal cysts includes a total excision, which is crucial due to the possibility of malignant transformation, especially in types I and IV. Complete extrahepatic bile duct cyst excision is usually performed via Roux-en-Y hepaticojejunostomy bilioenteric reconstruction.6 The management of choledochal cyst rupture with pseudocyst formation is similar in terms of the need for a complete excision of the cyst and pseudocyst. Prognosis is excellent in postoperative pediatric patients who undergo complete cyst resection; an 89% event-free rate and 90% 5-year overall survival rate are reported in these patients.6 Follow-up appointments are required as the risk of developing biliary malignancies remains high for years after the choledochal cyst excision.6
Conclusion
Choledochal cyst is an uncommon disease of childhood with cyst rupture being rare and pseudocyst formation following a rupture being even rarer. There are five types of choledochal cysts according to the Todani classification, with type I being the most common type. Complications can develop if the disease is not managed properly. Complete cyst excision is warranted due to the possibility of malignant formation. Outcome is excellent with complete excision but poor with malignant association and incomplete resection.
1. Altıntoprak F, Uzunoğlu MY, Dikicier E, Zengin I. Choledochal cysts—Classification, physiopathology, and clinical course. Integr Cancer Sci Therap. 2016;3(5):588-592. doi:10.15761/ICST.1000209
2. Arora M, Kaue L, Kaur J. Biliary pseudocyst: a rare complication of antenatal perforation of forme fruste choledochal cyst. J Indian Assoc Pediatr Surg. 2019;24(4):291-293. doi:10.4103/jiaps.JIAPS_212_18
3. Yasufuku M, Hisamatsu C, Nozaki N, Nishijima E. A very low-birth-weight infant with spontaneous perforation of a choledochal cyst and adjacent pseudocyst formation. J Pediatr Surg. 2012;47(7:E17-E19. doi:10.1016/j.jpedsurg.2012.03.055
4. Lipsett PA, Pitt HA, Colombani PM, Boitnott JK, Cameron, JL. Choledochal cyst disease. A changing pattern of presentation. Ann Surg. 1994;220(5):644-652. doi:10.1097/00000658-199411000-00007
5. Fumino S, Iwai N, Deguchi E, et al. Spontaneous rupture of choledochal cyst with pseudocyst formation-report on 2 cases and literature review. J Pediatr Surg. 2006;41(6):19-21. doi:10.1016/j.jpedsurg.2006.02.033
6. Soares KC, Arnaoutakis DJ, Kamel I, et al. Choledochal cysts: presentation, clinical differentiation, and management. J Am Coll Surg. 2014;216(6):1167-1180. doi:10.1016/j.jamcollsurg.2014.04.023