
Case in Point: Hypophosphatemic Rickets
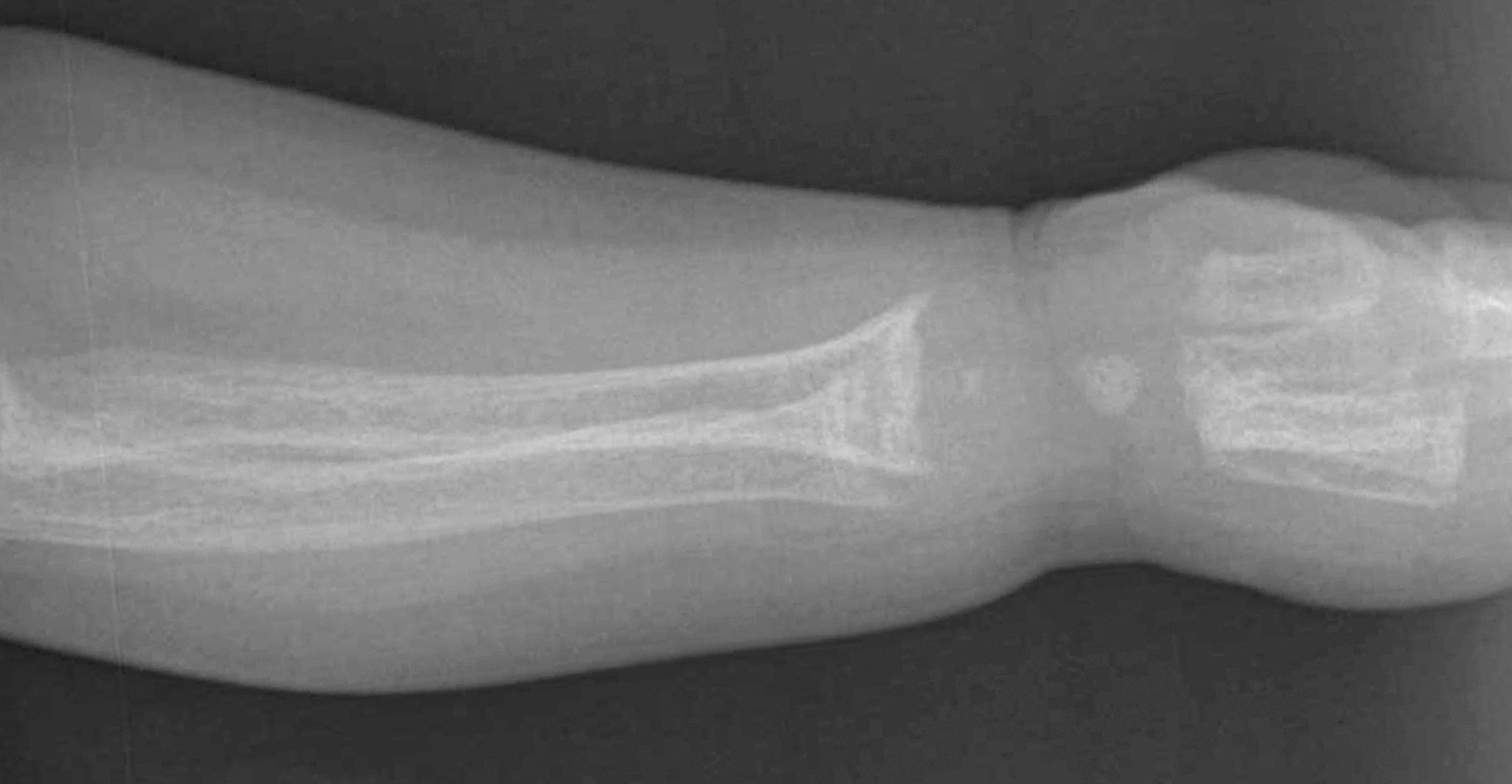
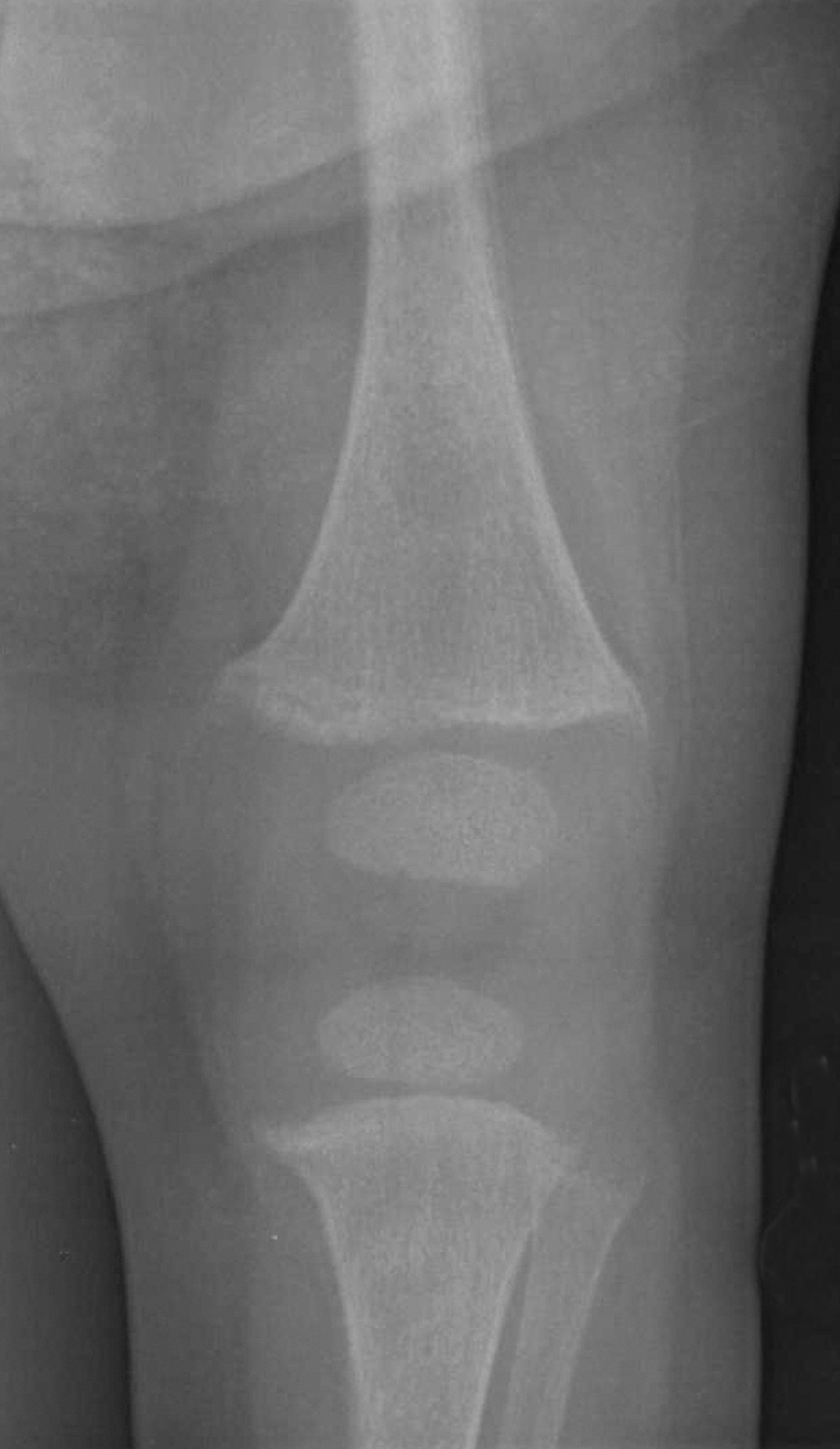
A dietary history revealed that the child had been fed a soy-based formula since early infancy because she had been unable to tolerate cow's milk. She drinks approximately 5 oz of soy-based formula every 2 to 3 hours and recently started to eat baby food.
A diagnosis of hypophosphatemic rickets was made. Therapy with vitamin D (calcitriol) and phosphorus supplementation was initiated.
The child's mother had rickets as a child, which left severe deformities. She was taking vitamin D and phosphorus supplements. The patient's 18-month-old sister had a history of delayed gross motor milestones and also had frontal bossing; however, a workup had never been done, nor had the child been treated.
Hypophosphatemic RicketsFamilial hypophosphatemic rickets is the most common nonnutritional form of rickets. The term "vitamin D-resistant rickets" was originally used to describe rickets that was refractory to treatment with vitamin D. It was later found that renal phosphate wasting and a low serum phosphate concentration were common factors in many refractory cases. This discovery led to the name "phosphate diabetes." This disorder now is more appropriately named "hereditary hypophosphatemic rickets." The usual mode of inheritance is X-linked dominant.
X-linked hypophosphatemic (XLH) rickets is the most common form of rickets in developed countries.1 One in 20,000 persons is affected.2,3 Other forms of hypophosphatemic rickets include autosomal dominant,4 autosomal recessive hereditary hypophosphatemic rickets with hypercalciuria,5 and oncogenic tumor-induced osteomalacia.1
Pathophysiology
The pathogenesis in XLH rickets involves a defect in proximal tubular reabsorption of phosphate and the conversion of 25-dihydroxy (OH) vitamin D to 1,25-dihydroxycholecalciferol (1,25[OH]2D). This is suggested by low 1,25(OH)2D levels despite hypophosphatemia.6,7 Studies indicate reduced activity of sodium-dependent phosphate transporter in the renal tubule.
Great strides have been made with the discovery of the gene responsible for XLH rickets on chromosome Xp22.1. This gene was named "PHEX" (Phosphate-regulating Endopeptidase on the X chromosome). Mutations in the PHEX gene result in under-expression of the sodium-phosphate co-transporter that is responsible for phosphate reabsorption in the proximal tubule.8,9 The PHEX gene inactivates hormone-like substances (called phosphatonins) that promote phosphate excretion and impair bone mineralization.10 In patients with XLH disease, these phosphatonins are not activated; consequently, they circulate in high concentrations, which leads to excessive phosphate excretion. Hormones recognized to be phosphatonins are fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein11 and frizzled-related protein 4.12
In autosomal dominant hypophosphatemic rickets, the FGF23 gene mutations on 12p13 interfere with its cleavage.13 Overproduction of FGF23 may be responsible for tumor-induced osteomalacia.14
Clinical FeaturesXLH rickets usually presents when a child begins to bear weight (ie, during the second or third year of life) and is manifested by the child's reluctance to walk. In XLH disease, the degree of involvement is less severe in heterozygous females. Nevertheless, no gender differences in disease severity were detected in the largest reported series of patients with XLH rickets.15 Affected patients grow slowly and have delayed dentition, dental abscesses, and early tooth decay. Males are affected more frequently than females.16 Both males and females may also have gross clinical findings of rickets; the lower extremities are more affected than the upper extremities. Enthesopathy (calcification of tendons, ligaments, and joint capsules) is a common finding.17
Laboratory FindingsHypophosphatemia and normal to slightly low calcium, normal to high parathyroid hormone, and increased alkaline phosphatase levels is a typical laboratory profile in patients with XLH rickets. The calcitriol( level is normal or slightly low (inappropriately, because hypophosphatemia stimulates vitamin D synthesis). Urinary loss of phosphate is always above reference ranges. Tubular reabsorption of phosphorus is 60% or less (normal is between 85% and 100%). Urinary calcium levels help distinguish this from hypercalciuric variants.
In all cases, radiography is the study of choice. However, there is no pathognomonic sign that differentiates hypophosphatemic rickets from any other type of rickets (Table). Histologically, osteomalacia and hypomineralized periosteocytic lesions are typically seen.
Distinguishing XLH From Other Causes
Autosomal dominant rickets manifests with hypophosphatemia and phosphaturia but minimal or no associated skeletal changes. Studies reveal that children present with phosphate wasting, rickets, and lower extremity deformity; some lose the renal phosphate-wasting defect after puberty. Adolescents and adults present with renal phosphate wasting, bone pain, weakness, and fractures but not with lower extremity deformity.4
In patients with autosomal recessive hypophosphatemic rickets, renal tubular phosphate handling is impaired but the response of a-hydroxylase to hypophosphatemia is intact. Thus, calcitriol levels are elevated, which increases intestinal calcium reabsorption and leads to hypercalciuria and osteopenic bones. Tumor-induced osteomalacia also has clinical findings similar to those of autosomal recessive hypophosphatemic rickets.
TreatmentThe goals of therapy are to improve growth, reduce the severity of bone disease, and minimize activity limitations. Phosphate and calcitriol are the mainstays of therapy. Phosphorus lowers the concentration of ionized calcium in plasma. Hypophosphatemia stimulates calcitriol synthesis; administration of phosphorus decreases calcitriol synthesis. There is secondary hyperparathyroidism from hypocalcemia. Increased levels of parathyroid hormone further aggravate phosphorus loss. Calcitriol administration is therefore necessary to increase the intestinal absorption of calcium and phosphorus and to prevent secondary hyperparathyroidism.
Therapy with calcitriol (Rocaltrol, Calcijex) is initiated at 15 to 20 ng/kg/d. The dosage is gradually increased over several months to 30 to 60 ng/kg/d. Phosphate salts are given at 0.5 to 1 g/24 h in a younger child and at 1 to 4 g/24 h in an older child in divided doses every 4 hours at least 5 times a day. Joulie's solution contains 30.4 mg of phosphorus per milliliter. Phospha-soda (Fleet) solution contains 127 mg of elemental phosphorus per milliliter. An older child may use Neutra-Phos-K powder (250 mg of elemental phosphorus per packet or capsule). The phosphorus dosage should be increased in 250- to 500-mg increments to a maximum of 3500 mg/d.
Affected children should be seen every 3 months. Healing typically starts in 6 to 8 weeks after the start of therapy. Monitor height; levels of serum calcium, phosphate, and alkaline phosphatase; and the urinary calcium- to-creatinine ratio (a ratio above 0.25 indicates hypercalciuria and the need to reduce the dose of 1,25[OH]2 D).18 Radiographs of the hand should be obtained to determine bone age and to monitor for the reappearance of rickets. Renal ultrasonography should be done to evaluate for nephrocalcinosis--a well-recognized complication of therapy.
Treatment is similar for the autosomal dominant form of rickets and for tumor-induced osteomalacia. The latter type is generally refractory to medical therapy; the defect in vitamin D metabolism is more severe and often results in low 1,25(OH)2D levels. The syndrome is cured when the tumor is removed.19-21
Hypophosphatemic rickets with hypercalciuria responds to phosphorus replacement alone.
Adjuvant TherapyDiuretics: Thiazide diuretics are administered to enhance calcium reabsorption and to reduce the risk of nephrocalcinosis. Amiloride( (Midamor) is used to counter hypokalemia as a result of thiazide therapy.
Growth hormone therapy: This has been tried in clinical trials in addition to the usual treatment. However, there are no current recommendations for this therapy.
Vitamin D analogs: Non-hypercalcemic analogs of vitamin D (ie, 24,25[OH]2D3) are being studied.16
SurgeryDespite medical therapy, some patients require surgical correction of severe bow defects. Because of the short half-life of 1,25(OH)2D and 1a-hydroxyvitamin D, it is not necessary to interrupt therapy before surgery. However, it is mandatory to monitor the urinary calcium-to-creatinine ratio. If the ratio exceeds 0.25, discontinue the 1,25(OH)2D for a few days.4
ComplicationsHypervitaminosis D occurs secondary to high doses of fat-soluble vitamin D and infrequent monitoring of serum and urine biochemistries.16 This complication is much less common today with the use of 1a- hydroxylated vitamin D metabolites.
Nephrocalcinosis, another complication, has been linked to excessive doses of calcitriol and to noncompliance with oral phosphate supplementation; the condition is associated with renal tubular acidosis.16,22,23 Hyperparathyroidism may develop from the stimulatory effect of phosphate on the parathyroid. Concomitant use of calcitriol dampens this effect.24
PrognosisAffected persons have a normal life span. With early diagnosis and good compliance with therapy, deformities can be minimized.
REFERENCES:
1. Rowe PS. The molecular background to hypophosphatemic rickets. Arch Dis Child. 2000;83:192-194.
2. DiMeglio LA, White KE, Econs MJ. Disorders of phosphate metabolism. Endocrinol Metab Clin North Am. 2000;29:591-609.
3. Tenenhouse HS, Murer H. Disorders of renal tubular phosphate transport. J Am Soc Nephrol. 2003;14:240-248.
4. Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab. 1997;82:674-681.
5. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78:193-201.
6. Chesney RW. Familial hypophosphatemia (vitamin D-resistant rickets associated with tumor). In: Behrman RE, Kliegman R, Jenson HB, eds. Nelson Textbook of Pediatrics. Philadelphia: Saunders; 2004:2345-2346.
7. Chesney RW. Oncogenous rickets (primary hypophosphatemic rickets associated with tumor). In: Behrman RE, Kliegman R, Jenson HB, eds. Nelson Textbook of Pediatrics. Philadelphia: Saunders; 2004:2346-2347.
8. Tenenhouse HS, Werner A, Biber J,et al. Renal Na(+)-phosphate cotransport in murine X-linked hypophosphatemic rickets. Molecular characterization. J Clin Invest. 1994;93:671-676.
9. Hruska KA, Rifas L, Cheng SL, et al. X-linked hypophosphatemic rickets and the murine Hyphomologue. Am J Physiol.1995;268:F357-F362.
10. Xiao ZS, Crenshaw M, Guo R, et al. Intrinsic mineralization defect in Hyp mouse osteoblasts. Am J Physiol. 1998;275:E700-E708.
11. Rowe PS, de Zoysa PA, Dong R, et al. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia.Genomics. 2000;67:54-68.
12. Berndt T, Craig TA, Bowe AE, et al. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest.2003;112:785-794.
13. White KE, Carn G, Lorenz-Depiereux B, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23.Kidney Int. 2001;60:2079-2086.
14. Shimada T, Mizutani S, Mutu T,et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia.Proc Natl Acad Sci U S A. 2001;98:6500-6505.
15. Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypophosphatemia: a search for gender, race, anticipation, or parent of origin effects on disease expression in children. J Clin Endocrinol Metab. 1996;81:4075-4080.
16. Carpenter TO. New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. Pediatr Clin North Am.1997;44:443-466.
17. Polisson RP, Martinez S, Khoury M, et al. Calcification of entheses associated with X-linked hypophosphatemic osteomalacia. N Engl J Med. 1985;313:1-6.
18. Kappy MS, Blizzard RM, Migeon CJ, eds. Wilkins the Diagnosis and Treatment of Endocrine Disorders in Childhood and Adolescence. Springfield, Ill: Charles C Thomas Publisher Ltd; 1994.
19. Agus ZS. Oncogenic hypophosphatemic osteomalacia. Kidney Int. 1983;24:113-123.
20. Salassa RM, Jowsey J, Arnaud CD. Hypophosphatemic osteomalacia associated with "nonendocrine" tumors. N Engl J Med. 1970;283:65-70.
21. Ryan EA, Reiss E. Oncogenous osteomalacia. Review of the world literature of 42 cases and report of two new cases.Am J Med. 1984;77:501-512.
22. Seikaly M, Browne R, Baum M. Nephrocalcinosis is associated with renal tubular acidosis in children with X-linked hypophosphatemia. Pediatrics. 1996;97:91-93.
23. Verge CF, Lam A, Simpson JM, et al. Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med.1991;325:1843-1848.
24. Bettinelli A, Bianchi ML, Mazzucchi E, et al. Acute effects of calcitriol and phosphate salts on mineral metabolism in children with hypophosphatemic rickets. J Pediatr. 1991;118:372-376.