
How the Times Have Changed: Kidney Disease
This Editorial is a personal reflection on an article from the Consultant archives and was written by a Consultant Editorial Board member.
Author:
James Matera, DO, FACOI
Schneiderman H. Bilateral flank masses due to adult poly cystic kidney disease. Consultant. 1990;30:43-44. https://www.consultant360.com/exclusives/Bulging-Bilateral-Flank-Masses.
I took a few minutes recently to review Dr Schneiderman’s article and image of bulging bilateral flank masses from 1990 in Consultant.1 When published, I was starting my career and knew at that time that nephrology was going to be my vocation. The first time I palpated enlarged flank masses in a patient with adult polycystic kidney disease (APKD) gave new meaning to physical diagnosis skills and stressed the anatomical differences we need to keep in mind. Identifying APKD has evolved since that time with advancement of the genetics, ultrasound diagnostic criterion, risk reduction strategies, and treatment options that have transcended conservative measures.
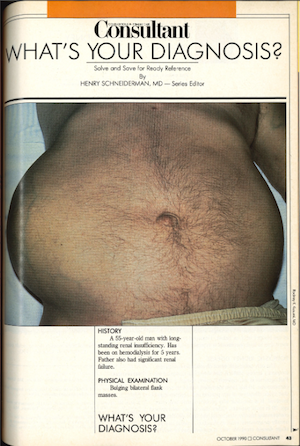
Confirmation of APKD requires imaging, which included ultrasonography, computed tomography (CT) urograms (much less useful today), CT scanning or magnetic resonance imaging (MRI), as physical examination is not enough. There are criteria for adults regarding ultrasound findings, which help in diagnosis.2 While we were aware in 1990 of the genetic basis of the disease, little was said about genetic testing or APKD1/APKD2 genes. About 85% of diagnosed cases contain Polycystin1 or PKD1 gene located on chromosome 16. The second gene, polycystin2 is less common and is located on chromosome 4. Less than 50% of patients will be identified due to the indolent and slow nature of the disease.3 Genetic testing can be done for patients with equivocal imaging or in a relative who may be a potential donor for transplantation.
Imaging remains the hallmark of diagnosis today and should be done in certain individuals with positive family history. It is also important to have genetic counseling prior to screening, as positive findings might lead to some adverse effects with insurability. Knowing the diagnosis, however, imparts knowledge concerning the diagnosis, appropriate family planning, the ability to detect and treat complications associated with the disease, reassurance of unaffected individuals, and appropriate selection of unaffected relatives as potential donors for kidney transplantation.
Ultrasound criteria for adults has been studied and based on cyst number and age4:
|
Age (years) |
Cyst Number |
Positive Predictive Value |
Unilateral/Bilateral |
|
15-39 |
3 |
100% |
Uni OR bilat |
|
40-59 |
2 |
90-100% |
In each kidney |
|
>60 |
4 |
~100% |
In each kidney |
Looking at the patient from 1990, the treatment was largely conservative once he was diagnosed. We need to look at epidemiology when discussing this aspect of care:
- ADPKD occurs in all races and has a reported prevalence of 1:400 to 1:1000.5
- ADPKD leads to 5% of patients who initiate dialysis annually in the United States.
- Patients can present with hypertension, hematuria, proteinuria, or renal insufficiency.
- Flank pain due to renal hemorrhage stones or infection are the most common symptoms.6
Treatment has been based on underlying causes like hypertension, stones, infections, pain, and bleeding. Use of renin angiotensin aldosterone antagonist inhibitors (RAASi) should be used in all patients with hypertension unless contraindicated.7 Genetic counselling is appropriate for family members, especially if diagnosis is in question. Lowering cardiovascular risk is also of utmost importance, as this is the leading cause of death in APKD patients. Data suggests that increasing fluid intake to 3 L/day helps suppresses urine osmolality and partially suppresses serum antidiuretic hormone (ADH) levels. A higher water intake may provide more complete suppression of ADH and cAMP.8 This finding led to studies involving the vasopressin receptor antagonist tolvaptan.
In REPRISE, among 1370 patients who were randomly assigned to tolvaptan or placebo, the change from baseline estimated glomerular filtration rate (eGFR) was lower among those assigned to tolvaptan compared with placebo. REPRISE studied patients with more advanced renal disease; patients aged 18 to 55 years had eGFRs 25 to 65 mL/min/1.73 m2, and those aged 56 to 65 years had eGFRs 25 to 44 mL/min/1.73 m2 with evidence of declining eGFR of at least 2 mL/min/1.73 m2.9 This led to the recent approval by the US Food and Drug Administration of tolvaptan as a potential option for our APKD patients.
Going back to the patient from 1990, the disease has remained the same, but our understanding and ability to alter the outcomes has changed and continues to change. This is consistent with the complex adaptive nature of medicine and medical therapy, and provides hope for future developments and reduction of the burden of end-stage renal disease in this genetic disease.10 Old dogs like me can even learn new tricks!
James J. Matera, DO, FACOI, is the Medical Director of Population Health at CentraState Medical Center in Freehold, New Jersey.
- Schneiderman H. Bilateral flanks masses due to adult polycystic kidney disease. Consultant. 1990;30:43-44. https://www.consultant360.com/exclusives/Bulging-Bilateral-Flank-Masses.
- Pei Y, Watnick T. Diagnosis and screening of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):140-152. doi: 10.1053/j.ackd.2009.12.001.
- Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350(2):151-164. doi:10.1056/NEJMra022161.
- Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205-212. doi:10.1681/ASN.2008050507.
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149-168. doi:10.1038/ki.2009.128.
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-1301. doi:10.1016/S0140-6736(07)60601-1.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009;20(9):1888-1893. doi:10.1681/ASN.2008080882.
- Torres VE, Bankir L, Grantham JJ. A case for water in the treatment of polycystic kidney disease. Clin J Am Soc Nephrol. 2009;4(6):1140-1150. doi:10.2215/CJN.00790209.
- Torres VE, Chapman AB, Devuyst O, et al; REPRISE Trial Investigators. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med. 2017;377(20):1930-1942. doi:10.1056/NEJMoa1710030.
- Martin CM. Complex adaptive systems approaches in health care—A slow but real emergence? J Eval in Clin Pract. 2018;24(1):266-268. doi:10.1111/jep.12878.