
Peer Reviewed
Neurosarcoidosis From Pituitary Expansion to Empty Sella
AFFILIATIONS:
1Internal Medicine Resident, Saint Peters University Hospital; Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ
2Medical Student, Rutgers University, Robert Wood Johnson Medical School, NJ
3Department of Internal Medicine, Saint Peters University Hospital / Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ
CITATION:
Thota G, Gribkova Y, Desai RN. Neurosarcoidosis from pituitary expansion to empty sella. Consultant. 2022;62(11):e10. doi:10.25270/con.2022.07.000015
Received January 31, 2022. Accepted April 27, 2022. Published online July 26, 2022.
DISCLOSURES:
The authors report no relevant financial relationships.
DISCLAIMER:
No written consent has been obtained from the patients as there is no patient identifiable data included in this case report.
CORRESPONDENCE:
Geethika Thota, MD, Saint Peter’s University Hospital, 254 Easton Ave., New Brunswick, NJ, 08901 (prathima.thota95@gmail.com)
A 68-year-old woman with a medical history of hypertension, hyperlipidemia, and hypothyroidism presented to the emergency department with a decrease in vision bilaterally, more on the right side, for 4 weeks prior to the presentation. She also reported an associated headache. The patient denied any nausea, vomiting, eye pain, eye discharge, recent viral illness, or changes in weight or appetite.
Physical examination. Upon presentation, the patient had a heart rate of 89 beats/min, a blood pressure of 145/89 mm Hg, a respiratory rate of 18 breaths/min, a temperature of 36.6 °C, and an oxygen saturation level of 100% on room air. Physical examination results were significant for bitemporal hemianopsia more on the right side than the left, decreased visual acuity more on the right than the left with normal extraocular muscle movement, and no focal neurological deficits.
Diagnostic testing. Laboratory values were as follows: white blood cells, 5.8 × 103/μL (reference, 4.0-11.0 × 103/μL); hemoglobin, 12.3 g/dL (reference, 12.0-16.0 g/dL); platelets, 239 × 103/μL (reference, 150-400 × 103/μL); erythrocyte sedimentation rate, 49 mm/hr (reference, 0-15 mm/hour); and angiotensin-converting enzyme (ACE) serum, 58 U/L (reference, 9-67 U/L).
A pituitary magnetic resonance imaging (MRI) study with and without contrast revealed the presence of infiltration of the pituitary gland (Figure 1), stalk and bilateral cavernous sinus (Figure 2), and tentorium. A computed tomography scan of the thorax with contrast showed the presence of shotty mediastinal and hilar nodes with one large subcarinal node measuring 2 cm.


Figure 1. MRI resolution of the sellar and suprasellar mass with an empty, expanded sella in 2021.
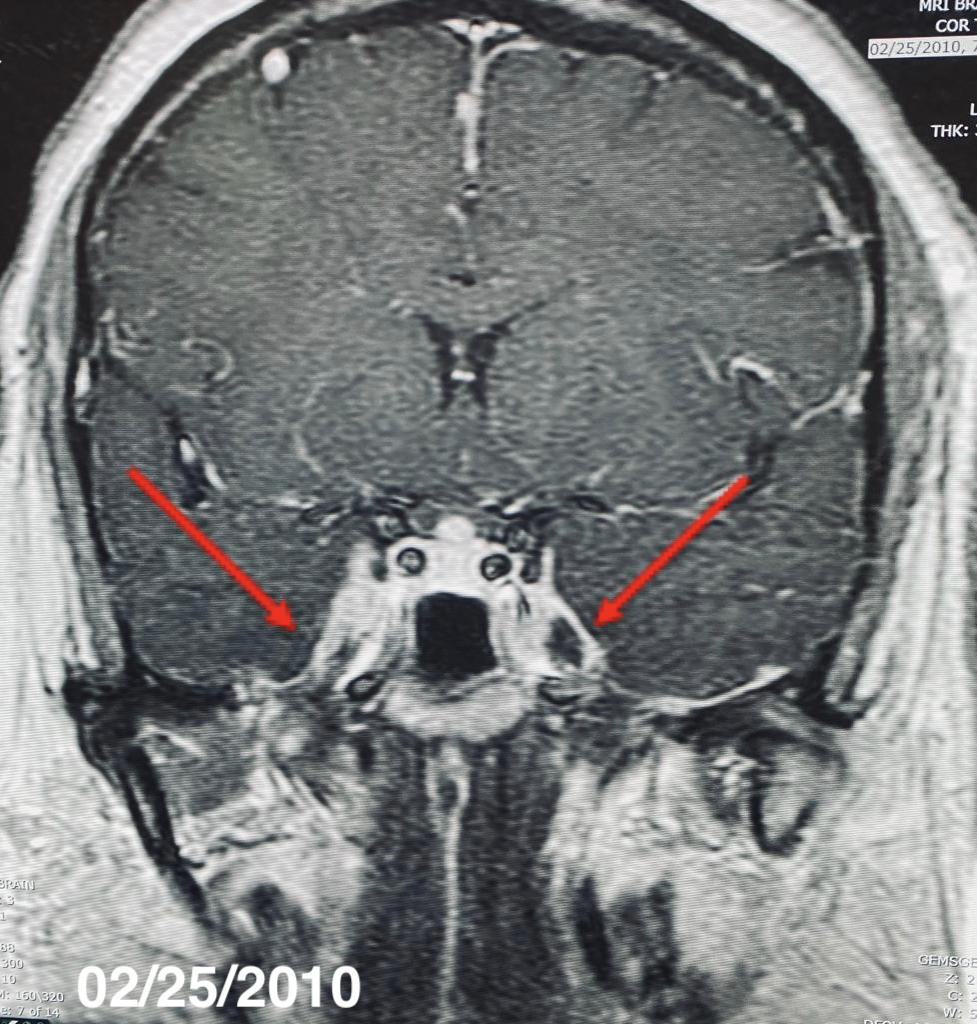
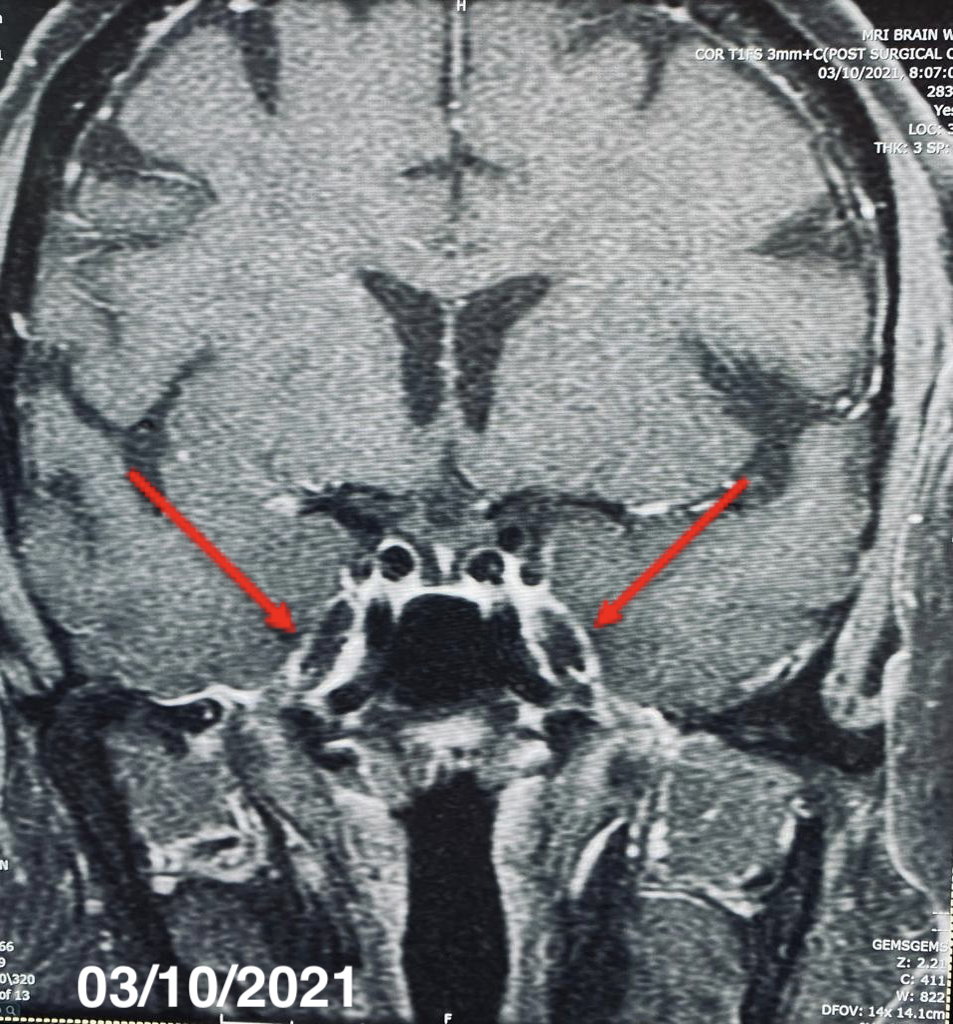
Figure 2. MRI showing regional dural thickening and enhancement along the cavernous sinuses bilaterally extending into the optic chiasms and superior orbital fissures, complete resolution in images of 2021.
The patient was started on intravenous dexamethasone, and a workup for sarcoidosis was conducted. A pituitary hormone analysis showed panhypopituitarism, with laboratory values as follows: serum follicle-stimulating hormone, less than 0.70 mIU/mL (reference, 1.5-12.4 mIU/mL); luteinizing hormone, less than 0.20 mIU/mL (reference, 1.37-9.00 mIU/mL); prolactin, 10.70 µg/dL (reference, 3.0-30.0 µg/dL); morning serum cortisol, less than 2 µg/dL (reference, 5-23 µg/dL); adrenocorticotropic hormone, less than 5 pg/mL (reference, 6-50 pg/mL); thyroid-stimulating hormone, 0.067 mIU/mL (reference, 0.465-4.680 mIU/mL); and free T4, 0.0039 µg/dL (reference, 0.0078-0.0219 µg/dL). Thyroid peroxidase and thyroglobulin antibodies test results were negative.
The patient underwent bronchial lavage, which showed histocytes and CD4/CD8 ratio of 0.8. The patient underwent lumbar puncture, with analysis of a cerebrospinal fluid, showing an ACE level of 9 U/L (reference, < 15 U/L). A diagnosis of hypopituitarism secondary to neurosarcoidosis was made, and the patient was started on prednisone, 60 mg/d gradually tapered to 7.5 mg/d. The patient has reported no recurrence of symptoms to date.
Patient Outcome. In 2022, 11 years after the diagnosis was made, the patient underwent a repeat MRI of the enhancing sellar and suprasellar mass, results of which showed ultimately an empty, expanded sella turcica. The patient has been clinically stable despite these changes. She is currently on a regimen of replacement of levothyroxine, 50 µg and prednisone, 7.5 mg daily. The patient remains symptom-free with complete recovery and remains clinically and biochemically euthyroid.
Discussion. Sarcoidosis is a multisystem granulomatous disorder characterized pathologically by the presence of noncaseating granulomas.1,2,6 Neurosarcoidosis (NS) is a heterogeneous disease resulting from nervous system involvement by sarcoid granulomas. Three criteria are usually required for definitive diagnosis of NS: clinical and radiologic manifestations, noncaseating sarcoid granulomas by histology, and no evidence of alternative disease.1,4
Sarcoidosis typically occurs in adults aged 20 to 40 years.1 About 5% to 15% of patients with sarcoidosis develop NS.1-3 Once NS is diagnosed, immunosuppression is the key to disease treatment, and corticosteroids play an important role along with other immunomodulatory drugs (eg, hydroxychloroquine, pentoxifylline, thalidomide, and infliximab) and immunosuppressive drugs (eg, methotrexate, azathioprine, cyclosporine, and cyclophosphamide).2,5,7 The long-term course of patients with NS remains unclear, with a mortality rate of 5% to 10%, as a direct consequence of its inflammatory state.8 The prognosis for patients with NS varies.2,9 Most patients have chronic progressive courses.2 MRI abnormalities usually improve with corticosteroid treatment, as seen in our case, but most endocrine defects are irreversible despite regression of the granulomatous process.2,7,8
Conclusion. Our case highlights the importance of considering NS high in the differential diagnosis of patients with known sarcoidosis who develop neurologic symptoms or in patients presenting de novo with a constellation of findings consistent with the disease. Our case also highlights the diagnostic evaluation and treatment of NS and the importance of appropriate follow-up care.
1. Voortman M, Drent M, Baughman RP. Management of neurosarcoidosis: a clinical challenge. Curr Opin Neurol. 2019;32(3):475-483. doi.10.1097/WCO.0000000000000684
2. Neurosarcoidosis. National Institute of Neurological Disorders and Stroke. Updated April 25, 2022. Accessed July 12, 2022. https://www.ninds.nih.gov/Disorders/All-Disorders/Neurosarcoidosis-Information-Page
3. Mubarik A, Hassan SM, Felix M, Muddassir S, Haq F. A confusing manifestation: a case report of neurosarcoidosis presenting with confusion. J Community Hosp Intern Med Perspect. 2018;8(6):363-367. doi.10.1080/20009666.2018.1536239
4. Segal BM. Neurosarcoidosis: diagnostic approaches and therapeutic strategies. Curr Opin Neurol. 2013;26(3):307-313. doi.10.1097/WCO.0b013e3283608459
5. Joseph FG, Scolding NJ. Neurosarcoidosis: a study of 30 new cases. J Neurol Neurosurg Psychiatry. 2009;80(3):297-304. doi.10.1136/jnnp.2008.151977
6. Thomas KW, Hunninghake GW. Sarcoidosis. JAMA. 2003;289(24):3300-3303. doi.10.1001/jama.289.24.3300
7. Chiang R, Marshall MC Jr, Rosman PM, Hotson G, Mannheimer E, Wallace EZ. Empty sella turcica in intracranial sarcoidosis. Pituitary insufficiency, primary polydipsia, and changing neuroradiologic findings. Arch Neurol. 1984;41(6):662-665. doi.10.1001/archneur.1984.04210080070017
8. Gerke AK. Morbidity and mortality in sarcoidosis. Curr Opin Pulm Med. 2014;20(5):472-8. doi:10.1097/MCP.0000000000000080
9. Joubert B, Chapelon-Abric C, Biard L, et al. Association of prognostic factors and immunosuppressive treatment with long-term outcomes in neurosarcoidosis. JAMA Neurol. 2017;74(11):1336-1344. doi.10.1001/jamaneurol.2017.2492