
Osteogenesis Imperfecta
Authors:
Chelsea King, BS; Laura Rosas, BBA; and Lynnette Mazur, MD, MPH
McGovern Medical School at the University of Texas Health Science Center at Houston
Citation:
King C, Rosas L, Mazur L. Osteogenesis imperfecta [published online April 16, 2018]. Consultant for Pediatricians.
An 8-year-old boy presented to our clinic for a well-child visit. He had no health concerns.
History. His medical history included a broken primary tooth at age 5 years, a dislocation of his wrist after falling off a couch at age 6 years, and a fracture of his left fifth finger after falling off a slide at age 7 years. The fracture had required open reduction and internal fixation. The review of systems was positive for easy bleeding and bruising and negative for hyperelastic skin, keloids, and poor wound healing.
Physical examination. Height was between the 25th and 50th percentiles at 124 cm, and weight was in the 75th percentile, at 26 kg. He had blue sclerae (Figure 1), was able to bend his thumb backward to touch his wrist (Figure 2), and was unable to fully extend his left fifth finger (Figure 3).



Diagnosis. The patient’s symptoms were concerning for osteogenesis imperfecta (OI) and Ehlers-Danlos syndrome (EDS) (Tables 1 and 2). The 2 conditions are associated with similar symptoms due to overlapping mutations of COL1A1, the gene that encodes type I collagen. Phenotypic similarities include blue-tinged sclerae, translucent skin, and hypermobile joints.1 However, fractures are more common with OI, while joint dislocations characterize EDS.2


Because of the negative family history for both disorders and the presence of common symptoms, genetic testing was performed. Results showed a heterozygous pathogenic variant in COL1A1, confirming a diagnosis of OI type I. No pathogenic variants were detected in COL1A2, the other gene that encodes collagen.
Discussion. Although 12 types of OI have been described, 4 main types are recognized (Table 3).3 High variability in genetic expressivity results in differences in severity between OI types and between patients with the same OI type. Although OI is inherited in an autosomal dominant pattern, most cases, such as that of our patient, arise through spontaneous mutation.4 The timing of presentation depends on the type of OI and can range from in utero to the first decade of life and sometimes to adulthood after OI is detected in an offspring. However, it is usually first diagnosed when a patient presents with recurrent fractures.5

The collagen defect in OI type I is a quantitative defect, and symptoms are less severe than in the other OI types with qualitative collagen defects. Therefore, the diagnosis of type 1 OI is often delayed; only a third of cases are diagnosed by age 1 year.4 Our patient’s broken tooth could be a presentation of dentinogenesis imperfecta type I (opalescent teeth or brittle teeth), a condition that can be part of OI type I.
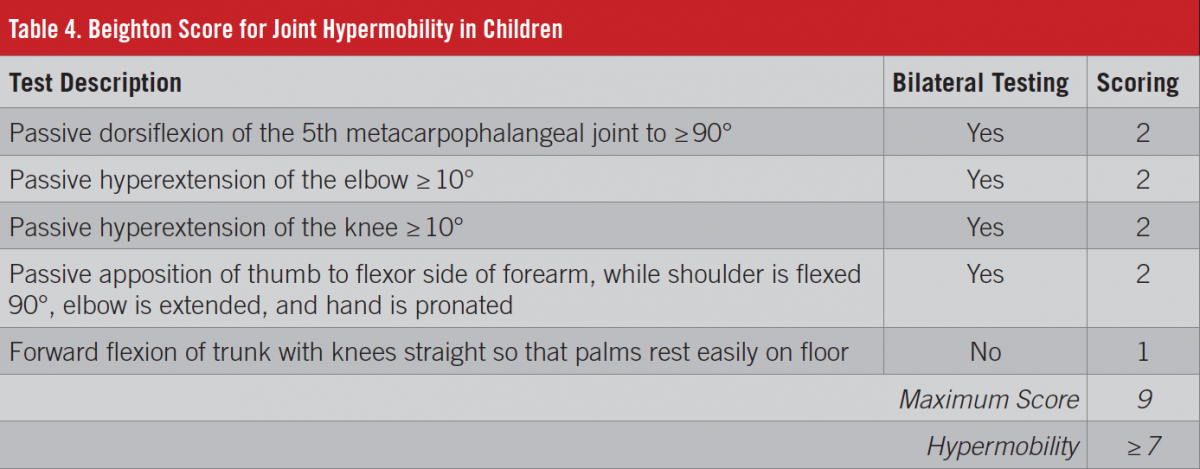
The 9-point Beighton score is used to evaluate the joint hypermobility that is often present in persons with OI (Table 4). Our patient’s score of 3 was in the normal range.6 Radiographs are usually indicated in patients presenting with fractures but are less sensitive in evaluating osteopenia,7 making these studies less reliable as a diagnostic tool for OI.
Treatment options for OI include lifestyle and diet changes. Adequate dietary intake of calcium, phosphorus, and vitamin D are essential, and supplementation of these nutrients may be indicated. Physiotherapy and regular joint-sparing exercise, particularly swimming, are vital to improving long-term quality of life.8 Bisphosphonate therapy is indicated for children with 2 or more long-bone fractures or 1 vertebral fracture in a year.
References:
- Colige A, Sieron AL, Li S-W, et al. Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet. 1999;65(2):308-317.
- Jones KL, Jones MC, Del Campo M. Osteogenesis imperfecta syndrome, type I. In: Jones KL, Jones MC, Del Campo M. Smith’s Recognizable Patterns of Human Malformation. 7th ed. Philadelphia, PA: Elsevier Saunders; 2013:634-638.
- Steiner RD, Adsit J, Basel D. COL1A1/2-related osteogenesis imperfecta. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK1295/. Published January 28, 2005. Updated February 14, 2013. Accessed April 12, 2018.
- Jones KL, Jones MC, Del Campo M. Osteogenesis imperfecta syndrome, type II. In: Jones KL, Jones MC, Del Campo M. Smith’s Recognizable Patterns of Human Malformation. 7th ed. Philadelphia, PA: Elsevier Saunders; 2013:638-642.
- Roughley PJ, Rauch F, Glorieux FH. Osteogenesis imperfecta—clinical and molecular diversity. Eur Cell Mater. 2003;5:41-47.
- Tofts LJ, Elliot EJ, Munns C, Pacey V, Sillence DO. The differential diagnosis of children with joint hypermobility: a review of the literature. 2009;7:1. Pediatr Rheumatol Online J. doi:10.1186/1546-0096-7-1.
- Sieber PR, Rommel FM, Theodoran CG, Russinko PJ, Woodward CA, Schimke L. The role of distal third radius dual energy x-ray absorptiometry (DXA) and central DXA in evaluating for osteopenia and osteoporosis in men receiving androgen deprivation therapy for prostate cancer. J Clin Densitom. 2012;15(3):351-354.
- Ramachandran M, Achan P, Jones DHA, Panchbhavi VK. Osteogenesis imperfecta treatment & management. Medscape. https://emedicine.medscape.com/article/1256726-treatment. Updated August 12, 2016. Accessed April 12, 2018.