
Peer Reviewed
Middle-Aged Woman With Severe Generalized Itchiness and Fatigability
Authors:
SIOBHAN MA, MD, PATRICK T. S. MA, MD, ALEXANDER K. C. LEUNG, MD, and ALEXANDER A. C. LEUNG, MD
Citation:
Ma S, Ma PTS, Leung AKC, Leung AAC. Middle-aged woman with severe generalized itchiness and fatigability. Consultant. August 2011:548-552.
HISTORY
A 50-year-old woman has a 6-month history of severe generalized itchiness and fatigability. There is no associated fever, abdominal pain, or joint pain. A cholecystectomy was performed 20 years earlier. She has no family history of hypercholesterolemia or liver disease.
PHYSICAL EXAMINATION
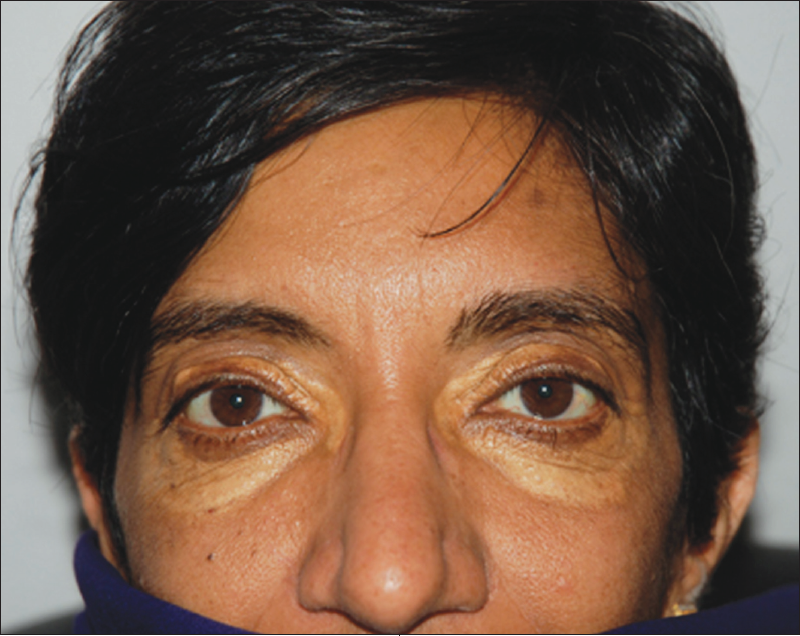
The patient has xanthelasma palpebrarum. Xanthomas are noted on the palms and elbows as well as the nape and left knee. The liver is 2 cm below the costal margin, firm, non-nodular, and nontender, with a span of 10 cm. There is no splenomegaly.
LABORATORY RESULTS
Laboratory results show markedly elevated levels of serum total cholesterol, 941.70 mg/dL; low-density lipoprotein (LDL), 890.73 mg/dL; alkaline phosphatase (ALP), 1301 U/L; alanine aminotransferase (ALT), 138 U/L; albumin, 2.2 g/L; and direct bilirubin, 2.40 mg/dL. The antimitochondrial antibody (AMA) titer is greater than 1:640.
The clinical and laboratory findings suggested primary biliary cirrhosis (PBC). Results of abdominal ultrasonography, CT, and MRI were normal. The patient was treated with ursodiol (15 mg/kg/d in 3 divided doses).
WHAT'S YOUR DIAGNOSIS? >>
ANSWER: PRIMARY BILIARY CIRRHOSIS
BILIARY CIRRHOSIS: AN OVERVIEW
Biliary cirrhosis is an autoimmune liver disorder that generally results from injury to or prolonged obstruction of either the intrahepatic or extrahepatic biliary system. It also may be caused by impaired biliary excretion or destruction of small intrahepatic bile ducts and canals of Hering with progressive fibrosis.1,2
PBC is characterized by chronic inflammation as well as fibrous obliteration of the intrahepatic bile ducts; the disease is progressive and can lead to liver damage and ultimately liver failure. In contrast, secondary biliary cirrhosis is defined as prolonged obstruction of the larger extrahepatic ducts. PBC affects all races, although it is most common in whites.3 The prevalence ranges from 19 to 240 cases per million.4 The female to male ratio is 10 to 1.3 Ninety percent of affected women are between 35 and 60 years of age.2
PATHOGENESIS
Although the precise cause is unknown, both environmental and genetic factors may play a role in the pathogenesis of PBC.5 The condition is associated with autoimmune disorders such as the CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia), Sjögren syndrome, autoimmune thyroiditis, rheumatoid arthritis, pernicious anemia, type 1 diabetes mellitus, type 1 renal tubular acidosis, and IgA deficiency. PBC has also been reported as an extraintestinal manifestation of celiac disease.4
IgG AMA is detected in more than 90% of patients with PBC.2 These antibodies are directed against the pyruvate dehydrogenase and 2-oxo-acid enzymes in the mitochondria. In addition, a number of class II major histocompatibility loci, including DR8, have been observed in patients with PBC.
CLINICAL FEATURES
About 40% of patients are asymptomatic at the time of diagnosis. Pruritus and fatigue are the usual presenting symptoms. Pruritus can be generalized or isolated to the palms and soles2; typically the onset is insidious. The severity of fatigue is independent of the extent of the hepatic disease. Some patients may have anorexia, nausea, and right upper quadrant abdominal pain.

As the disease progresses, jaundice develops. Affected patients may have hepatomegaly, splenomegaly, hyperpigmentation, and clubbing of the fingers. Steatorrhea and malabsorption of lipid-soluble vitamins may occur because of deficiency of bile salts in the intestine and pancreatic exocrine inefficiency.6 Subcutaneous deposition of lipoprotein around the eyes (xanthelasma palpebrarum) and over joints and tendons (xanthoma) (Figure) may also occur. Other clinical features include bone tenderness, glossitis, dermatitis, ascites, and ecchymosis.2

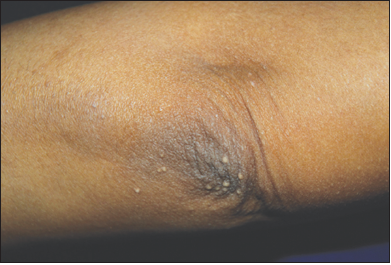
Figure. Xanthomas, seen here on the patient’s palms and elbows, can be manifestations of primary biliary cirrhosis.
DIAGNOSIS
Blood cell counts are usually normal in the early stages of the disease.2 PBC is typically diagnosed at a presymptomatic stage based on the results of a routine liver biochemistry screening that show marked elevation of serum ALP (usually 3 to 4 times the upper limit of normal), ALT, aspartate aminotransferase (AST), and gamma-glutamyl transpeptidase. The diagnosis can be confirmed by the detection of AMA in the serum; this test is fairly specific and sensitive.6
Patients with PBC commonly have elevated levels of serum bile acid and serum IgM. Hypoalbuminemia, hyperbilirubinemia, and a prolonged prothrombin time/international normalized ratio are associated with poor prognosis. Tests for rheumatoid factor, anti–smooth muscle antibodies, antinuclear antibodies, and thyroid antibodies may be positive. An elevated antitransglutaminase antibody titer indicates associated celiac disease.
A cholestatic pattern of liver disease and markedly elevated cholesterol levels are hallmark features of PBC on laboratory blood work. The serum cholesterol level is high in more than half of patients and may even exceed 1003.86 mg/dL.7,8 LDL and very low-density lipoprotein levels are typically only mildly elevated, whereas high-density lipoprotein (HDL) levels are markedly increased.9
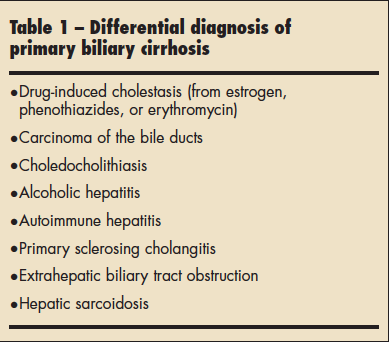
The differential diagnosis of PBC is shown in Table 1.2 Ultrasonography of the abdomen may help exclude extrahepatic obstruction. Liver biopsy helps establish the diagnosis and determine the stage of disease (Table 2). Serum bilirubin levels can be used to monitor disease progression.2

COMPLICATIONS
Malabsorption of fat-soluble vitamins is a common complication. Osteomalacia may result from decreased vitamin D absorption. This may predispose affected patients, especially postmenopausal women, to osteoporosis.2 Patients with a high level of fatigue may become emotionally stressed and depressed.10
Hepatic encephalopathy and complications from portal hypertension, including variceal hemorrhage, ascites, and hepatorenal syndrome, may occur.11 Hepatocellular carcinoma is more common in men and rare in women.
Despite the strikingly elevated cholesterol levels in patients with PBC, the risk of atherosclerotic death is not increased.12 High HDL levels may be protective against atherosclerotic disease in these patients.
TREATMENT
Management of PBC is mainly symptomatic. Ursodiol or ursodeoxycholic acid (UDCA) promotes secretion of endogenous bile acids and decreases cytokine production. It is used to lower serum levels of ALP, ALT, and AST and to alleviate pruritus.13 UDCA is most effective in patients with early disease.14 Side effects include diarrhea and rashes.
It remains unclear whether UDCA delays disease progression. Studies suggest no mortality benefit or reduction in liver transplantation with UDCA.15,16 Patients with advanced PBC and complications of portal hypertension do not benefit from UDCA and should be considered for liver transplantation. Recurrence of PBC after liver transplantation is uncommon.
Cholestyramine, an oral bile salt-sequestering resin, can be used to reduce the intensity of pruritus and the degree of hypercholesterolemia.2 Plasmapheresis may be considered in patients with refractory pruritus.
Steatorrhea may be reduced with a low-fat diet. Supplementation with fat-soluble vitamins (vitamins A, D, E, and K) should also be given.
Periodic screening for osteomalacia and osteoporosis with bone densitometry is important in patients
who have PBC. Patients with osteoporosis may possibly benefit from vitamin D, calcium supplements, and bisphosphonate therapy.2
Dr S. Ma is a resident in internal medicine at the Royal College of Surgeons in Ireland.
Dr P. T. S. Ma is clinical assistant professor of medicine.
Dr A. K. C. Leung is clinical professor of pediatrics both at the University of Calgary in Alberta.
Dr A. A. C. Leung is a clinical scholar in medicine at the University of Calgary in Alberta and a research fellow at the Brigham and Women’s Hospital and Harvard Medical School in Boston, Massachusetts.
References
- Washington MK. Autoimmune liver disease: overlap and outliers. Mod Pathol. 2007;20(suppl 1):S15-S30.
- Chung RT, Podolsky DK. Cirrhosis and its complications. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison’s Principles of Internal Medicine. 16th ed. New York: McGraw-Hill; 2005:1857-1873.
- Jones DE. Pathogenesis of primary biliary cirrhosis. Gut. 2007;56:1615-1624.
- Duggan JM, Duggan AE. Systematic review: the liver in coeliac disease. Aliment Pharmacol Ther. 2005;21:515-518.
- Lazaridis KN, Juran BD, Boe GM, et al. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46:785-792.
- Leuschner U. Primary biliary cirrhosis—presentation and diagnosis. Clin Liver Dis. 2003;7:741-758.
- Sherlock S, Scheuer PJ. The presentation and diagnosis of 100 patients with primary biliary cirrhosis. N Engl J Med. 1973;289:674-678.
- Iannuzzi C, Cozzolino G, Negro G. Elective cholecystectomy in selected cirrhotic patients. Acta Chir Belg. 1993;93:147-150.
- Jahn CE, Schaefer EJ, Taam LA, et al. Lipoprotein abnormalities in primary biliary cirrhosis. Association with hepatic lipase inhibition as well as altered cholesterol esterification. Gastroenterology. 1985;89:1266-1278.
- Blackburn P, Freeston M, Baker CR, et al. The role of psychological factors in the fatigue of primary biliary cirrhosis. Liver Int. 2007;27:654-661.
- Ishibashi H, Komori A, Shimoda S, Gershwin ME. Guidelines for therapy of autoimmune liver disease. Semin Liver Dis. 2007;27:214-226.
- Crippin JS, Lindor KD, Jorgensen R, et al. Hypercholesterolemia and atherosclerosis in primary biliary cirrhosis: what is the risk? Hepatology. 1992;15:858-862.
- Poupon R, Chrétien Y, Poupon RE, et al. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet. 1987;1:834-836.
- Corpechot C, Carrat F, Bahr A, et al. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology. 2005;128:297-303.
- Gong Y, Huang Z, Christensen E, Gluud C. Ursodeoxycholic acid for patients with primary biliary cirrhosis: an updated systematic review and meta-analysis of randomized clinical trials using Bayesian approach as sensitivity analyses. Am J Gastroenterol. 2007;102:1799-1807.
- Burroughs AK, Leandro G, Goulis J. Urso- deooxycholic acid for primary biliary cirrhosis. J Hepatol. 2001;34:352-353.
- Metcalf JV, Bhopal RS, Gray J, et al. Incidence and prevalence of primary biliary cirrhosis in the city of Newcastle upon Tyne, England. Int J Epidemiol. 1997;26:830-836.