
Peer Reviewed
What Explains This Child’s Skin and Hair Abnormalities?
Authors:
Alexander K. C. Leung, MD; Kin Fon Leong, MD; and Joseph M. Lam, MD
Citation:
Leung AKC, Leong KF, Lam JM. What explains this child’s skin and hair abnormalities? Consultant. 2018;58(6):e194.
A 4-day-old Malay boy presented with generalized erythematous pruritic scaly plaques and sparse hair. He had been born at 39 weeks of gestation to a 28-year-old mother following an uncomplicated pregnancy and spontaneous vaginal delivery. The Apgar scores were 5 and 8 at 1 and 5 minutes, respectively. Birth weight was 2.65 kg, length was 48 cm, and head circumference was 33 cm. The parents were nonconsanguineous. There was no family history of a similar dermatosis. His 33-year-old father had allergic rhinitis, and his 8-year-old sister had atopic dermatitis and asthma.
Physical examination revealed generalized erythroderma and desquamation (Figures 1 and 2). The scalp was covered with thick, adherent, scaly plaques, and the hair was very short, brittle, and sparse (Figure 3). The eyelashes and eyebrows were sparse. No ectropion or eclabium was noted. The rest of the physical examination findings were normal.


Figures 1 and 2. Physical examination of the neonate revealed generalized erythroderma and desquamation.
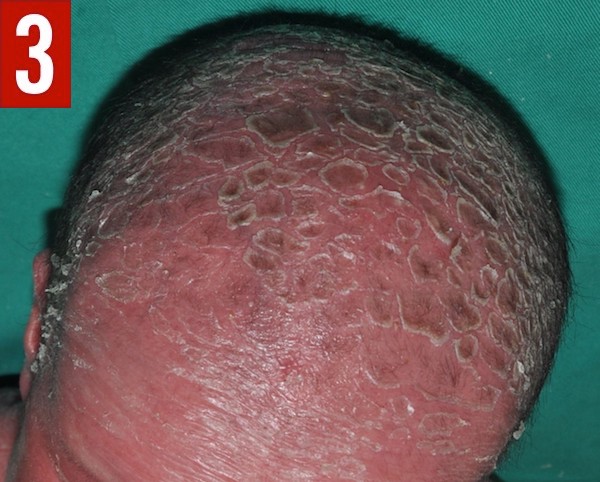
Figure 3. The neonate’s scalp was covered with thick, adherent, scaly plaques, and the hair was very short, brittle, and sparse.
Results of laboratory studies at presentation were remarkable for a serum immunoglobulin E (IgE) level of 4200 IU/mL (reference range, 0-100 IU/mL) and an absolute eosinophil count of 14,000/µL (reference range, 30-350/µL).
At 9 months of age, trichoscopic examination (Figure 4) and light microscopy (Figure 5) of the scalp hair showed trichorrhexis invaginata (bamboo hair). A skin biopsy at the same time showed acanthosis, parakeratosis, and psoriasiform epidermal hyperplasia. Immunohistochemistry staining of the skin biopsy specimen using specific antibodies showed a complete absence of lymphoepithelial Kazal-type related inhibitor (LEKTI) at the upper epidermis.
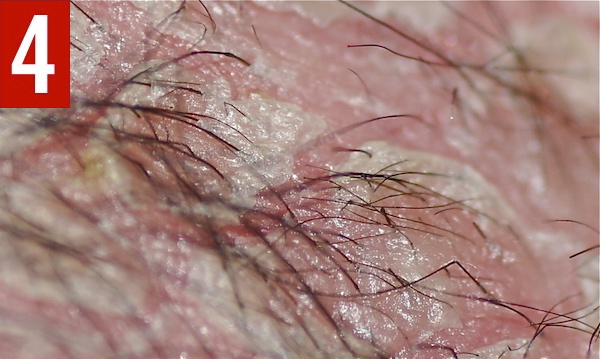
Figure 4. Trichoscopic examination of the scalp showed trichorrhexis invaginata.

Figure 5. Microscopic examination of the scalp hair showed trichorrhexis invaginata.
During the first year of life, the child had atopic dermatitis, recurrent infections (impetigo, cellulitis, gastroenteritis, otitis media, pneumonia), hypernatremic dehydration, and failure to thrive (body weight was 7.3 kg at 1 year). The erythroderma disappeared with time and progressed to serpiginous, pruriginous, erythematous plaques with double-edged scales at the periphery at 5 years of age.
Answer: Netherton Syndrome
Netherton syndrome, also known as Comèl-Netherton syndrome, is a rare disorder of cornification characterized by the triad of congenital ichthyosiform erythroderma/ichthyosis linearis circumflexa, trichorrhexis invaginata (bamboo hair), and an atopic diathesis.1 The term ichthyosis linearis circumflexa was first used by Comèl in 1949.2 In 1958, Netherton reported on a young girl with generalized erythematous scaly dermatitis and bamboo-like nodes in the sparse fragile hair. The disorder now bears their names.3
EPIDEMIOLOGY
The incidence of Netherton syndrome is 1 per 100,000 to 200,000 births.4 Both sexes are equally affected.4 All ethnic and racial groups are at risk.4
PATHOGENESIS
Netherton syndrome is inherited in an autosomal recessive fashion.4,5 The syndrome is caused by germline mutations of both copies of the serine protease inhibitor of Kazal type 5 gene (SPINK5) located on chromosome 5, long arm, band 32.5-7 The gene encodes the serine protease inhibitor LEKTI, which is predominately expressed in the lamellar granules of mucosal and epithelial surfaces and in the thymus.7-10 More than 70 distinct SPINK5 mutations specific to an individual or family have been described.8 Each SPINK5 mutation (eg, deletion, insertion, nonsense) leads to a different length of LEKTI protein, which accounts for the difference in severity of the disorder.6 Mutations in SPINK5 result in an unopposed and uncontrolled activity of epidermal proteases, leading to premature and increased desquamation of the stratum corneum and impairment of the skin barrier function.10 Impairment of the skin barrier function may lead to increased susceptibility to infection and aggravation of skin lesions.5,11
HISTOPATHOLOGY
Histopathologic examination of the lesion shows psoriasiform epidermal hyperplasia, acanthosis, parakeratosis, dyskeratosis, clear cells in the stratum corneum, dilated blood vessels in the superficial dermis, perivascular neutrophilic/eosinophilic infiltrate in the dermis, and subcorneal/intracorneal splitting.8,10 Immunostaining for LEKTI is negative.8
NEXT: Clinical Manifestations
CLINICAL MANIFESTATIONS
Netherton syndrome is characterized by the triad of congenital ichthyosiform erythroderma/ichthyosis linearis circumflexa, trichorrhexis invaginata (bamboo hair or ball-and-socket hair-shaft deformity), and an atopic diathesis.1,4,12 Patients with Netherton syndrome may show all or some of these features with varying degrees of severity.
Congenital ichthyosiform erythroderma is often present at birth or during the first weeks of life and manifests as generalized reddening of the skin (erythroderma) and dry fine scales (ichthyosis),13 as illustrated in the present case. Congenital erythroderma is a hallmark of Netherton syndrome.1 Congenital alopecia may be present.8 Some infants are born with a collodion membrane.1 With time, the erythroderma often progresses gradually into ichthyosis linearis circumflexa, characterized by migratory, circinate, polycyclic, serpiginous, pruriginous, erythematous plaques with double-edged scales at the periphery (Figure 6).1,14 Ichthyosis linearis circumflexa is another hallmark of Netherton syndrome and usually occurs after 2 years of age. The lesions are pruritic and wax and wane throughout life.1,4,14 Ichthyosis linearis circumflexa tends to last longer in adults than in children. Sites of predilection include the trunk and extremities.10 Lichenification of the antecubital and popliteal fossae is common.1,10,14 Taut facial skin and ectropion can develop in older children with severe disease.
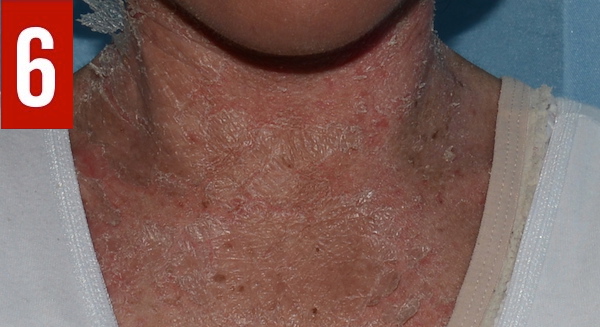
Figure 6. The erythroderma of Netherton syndrome often progresses gradually into ichthyosis linearis circumflexa, as shown in this patient’s case.
The hair is typically dry, lusterless, short, sparse, thin, beaded, brittle, spiky, fragile, and slow-growing.10,15-17 Because hairs in normal infants may also be sparse in the first few months of life, the hair shaft abnormality that is characteristic of Netherton syndrome can easily be overlooked. Trichorrhexis invaginata of scalp hair, eyelashes, and eyebrows, due to the invagination of the distal part of the hair shaft into the proximal part, is pathognomonic for Netherton syndrome.17,18 When hair breaks at the point of invagination, the appearance simulates a matchstick or golf tee.14-16 Hair shaft abnormalities usually do not develop until later in infancy or early childhood and are best visualized with a dermatoscope (trichoscopy) or under microscopy (trichogram).4 It is not unusual for hundreds of hairs to be examined before a hair with trichorrhexis invaginata is found.18 The hair-shaft abnormality is best visualized in eyebrow hair and in hair over pressure areas such as the occipital and temporal scalp.1,15,16,18,19 Some patients may have normal-looking hair, with the hair-shaft abnormality only noted when examined under trichoscopy or microscopy (subclinical trichorrhexis invaginata). Older patients may lose eyebrows and eyelashes together. Some patients may have trichorrhexis and pili torti.8
Atopic manifestations include atopic dermatitis, eczematous dermatitis with periorificial accentuation, urticaria, angioedema, asthma, allergic rhinitis, anaphylactic reactions to food (especially to nuts, eggs, and fish), hypereosinophilia, and elevation of serum IgE.1,4,9,10,17
Other inconsistent features that may be present include heat intolerance, mild mental retardation, growth retardation, hypoalbuminemia, immunodeficiency, enteropathy with villous atrophy, and exocrine pancreatic insufficiency.7,9,20
NEXT: Diagnosis
DIAGNOSIS
A high index of suspicion is necessary. The following features support the diagnosis of Netherton syndrome: scaling erythroderma in early infancy or ichthyosis linearis circumflexa in later childhood; trichorrhexis invaginata; atopic diathesis; and a family history of Netherton syndrome.8 In this regard, recalcitrant eczematous dermatitis that does not respond to topical corticosteroid therapy increases the possibility of Netherton syndrome.8 Atopic diathesis, although present in most cases, is not invariably present.21,22 The diagnosis of Netherton syndrome can be confirmed, if necessary, by identification of a germline SPINK5 mutation by DNA sequencing.8 The cost of performing DNA sequencing analysis, however, limits its routine use in clinical practice.16
DIFFERENTIAL DIAGNOSIS
Differential diagnosis includes autosomal recessive congenital ichthyosis; exfoliative (epidermolytic) ichthyosis; staphylococcal scalded skin syndrome; atopic dermatitis; psoriasis; seborrheic dermatitis; acrodermatitis enteropathica; generalized (type B) inflammatory peeling skin syndrome; and severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome.4,8,12
Autosomal recessive congenital ichthyosis is an heterogeneous group of disorders of keratinization with 4 major subtypes, namely, harlequin ichthyosis, lamellar ichthyosis, congenital ichthyosiform erythroderma, and pleomorphic ichthyosis.23,24 Infants with the severe forms are born enveloped in a collodion membrane that is replaced during the first weeks of life by the definitive phenotype. Harlequin ichthyosis is characterized by thick, adherent, and diamond-shaped armor-like scales, deep cutaneous cracks and fissures, contractures of digits, restricted movement, severe ectropion, fish-mouth-like eclabium, and flattened nose and ears. Patients with lamellar ichthyosis have large, dark brown, plate-like scales that are centrally adherent with raised edges and resemble a suit of armor. Tautness of the facial skin might result in eversion of the eyelids (ectropion) and lips (eclabium). Patients with congenital ichthyosiform erythroderma present with pronounced erythroderma and fine white scales, which distinguish this condition from other forms of ichthyosis. Pleomorphic ichthyosis, the mildest form of autosomal recessive congenital ichthyosis, is characterized by marked hyperkeratosis at birth followed by spontaneous improvement during infancy and mild cutaneous symptoms and signs thereafter.23 The absence of hair-shaft abnormality and atopic diathesis in autosomal recessive congenital ichthyosis distinguishes this condition from Netherton syndrome.
Staphylococcal scalded skin syndrome typically presents with tender erythroderma, bullae, and desquamation with a scalded appearance, especially in friction zones.25 Other features include periorificial scabs/crusting and positive Nikolsky sign.25 The erythroderma and desquamation may mimic that of congenital erythroderma, which is a hallmark of Netherton syndrome. However, the erythroderma and desquamation seen in staphylococcal scalded skin syndrome are not congenital, and the Nikolsky sign is negative in Netherton syndrome. In addition, trichorrhexis invaginata and atopic diathesis are not features of staphylococcal scalded skin syndrome.
NEXT: Differential Diagnosis (Continued)
Atopic dermatitis can be misdiagnosed as Netherton syndrome due to the presence of eczematous lesions and a personal or family history of atopy in Netherton syndrome. However, in atopic dermatitis, congenital ichthyosiform erythroderma/ichthyosis linearis circumflexa and trichorrhexis invaginata are absent.
Psoriasis is characterized by sharply demarcated, erythematous, round or oval plaques with loosely adherent silvery white micaceous scales.26 Removal of the scales results in fine punctate bleeding (Auspitz sign).26 The lesions are usually symmetrically distributed and pruritic. New lesions may form at the site of trauma (Koebner phenomenon).26 Most patients eventually develop nail involvement, which includes pitting, discoloration, onycholysis, or onychodystrophy.26 Congenital psoriasis and Netherton syndrome can also present as neonatal erythroderma and scaling with overlapping features of recurrent skin infections and failure to thrive.6 Cases of Netherton syndrome mimicking psoriasis have been described.6,12 In Netherton syndrome, the Auspitz sign and the Koebner phenomenon are negative, and nail involvement is not a feature. On the other hand, patients with psoriasis do not have trichorrhexis invaginata and the atopic diathesis seen in patients with Netherton syndrome.
Seborrheic dermatitis is a common chronic inflammatory skin disease characterized by erythema and greasy scales affecting areas rich in sebaceous glands.27 Infants with seborrheic dermatitis often present with focal or diffuse scaling and crusting of the scalp. Erythematous or salmon-colored sharply demarcated patches with yellow-white scales may involve the face, postauricular areas, trunk, and intertriginous and flexural areas of the body.27 In the diaper area, infantile seborrheic dermatitis presents as a sharply demarcated, erythematous, scaly eruption with a tendency to coalesce, resulting in the formation of a large confluent lesion. Pruritus is characteristically absent. In seborrheic dermatitis, the scaling is typically greasier, and sites of predilection are different from those of Netherton syndrome.8 The absence of trichorrhexis invaginata and atopic diathesis supports the diagnosis of seborrheic dermatitis.
Acrodermatitis enteropathica, caused by inherited or acquired zinc deficiency, is characterized by acral and periorificial dermatitis, diarrhea, failure to thrive, and alopecia.28 Classically, cutaneous lesions present with symmetrical, well-demarcated, erythematous, scaly, crusted, and variably eroded plaques. The eruptions may appear eczematous, psoriasiform, and, less commonly, vesiculobullous. The skin eruption, diarrhea, and failure to thrive may mimic the signs of in Netherton syndrome. Patients with acrodermatitis enteropathica do not have trichorrhexis invaginata, and the incidence of atopic diathesis is not increased.
Peeling skin syndrome is a heterogeneous group of autosomal recessive disorders characterized by superficial painless peeling with blistering of the skin without mucosal involvement. Two major forms of peeling skin syndrome are recognized, namely, acral peeling skin syndrome and generalized peeling skin syndrome. The latter is subclassified into generalized noninflammatory (type A) peeling skin syndrome and generalized (type B) inflammatory peeling skin syndrome. Patients with generalized (type B) inflammatory peeling skin syndrome typically present with ichthyosiform erythroderma at birth, superficial skin peeling, severe pruritus, food allergies, urticaria, angioedema, asthma, recurrent skin infections, and failure to thrive.8 These features are also commonly seen in patients with Netherton syndrome. The absence of a hair-shaft abnormality favors the diagnosis of generalized (type B) inflammatory peeling skin syndrome.
SAM syndrome is an autosomal recessive inherited genodermatosis caused by homozygous mutations in the desmoglein 1 gene (DSG1).29,30 The syndrome is characterized by congenital erythroderma, scaling, severe dermatitis, palmoplantar keratoderma, skin erosions, multiple food allergies, elevated IgE levels, recurrent infections, malabsorption, metabolic wasting, and failure to thrive.29,30 Affected patients have hard, curly hair but not trichorrhexis invaginata, which helps to distinguish this condition from Netherton syndrome.
NEXT: Complications & Prognosis
COMPLICATIONS
Recurrent infections, especially bacterial infections of the skin, are common and occur in at least 30% of affected patients.9 In one study, recurrent infections, either cutaneous or systemic, occurred in almost all affected patients.9 Other complications include hypernatremic dehydration, hypothermia/altered thermoregulation due to excessive transepidermal fluid loss, aminoaciduria, and failure to thrive.7,9,10,14,31,32 The pruritic skin lesions may cause irritability and insomnia.14 Affected individuals are at increased risk for early-onset cutaneous neoplasms, notably squamous cell carcinoma and basal cell carcinoma.4,33,34 Other rare complications include cardiomyopathy, pulmonary hypertension, renal vein thrombosis, hypothyroidism, intracranial hemorrhage, rickets, and pancreatitis.20,35-37 Due to their skin barrier dysfunction, patients with Netherton syndrome are also at risk for increased percutaneous absorption of topical substances and medicines.38
PROGNOSIS
The mortality rate is approximately 20% in the first year of life.8 Mortality is mainly due to systemic sepsis and hypernatremic dehydration.14 The skin condition usually improves with age but tends to fluctuate.14
NEXT: Management
MANAGEMENT
Treatment of patients with Netherton syndrome is challenging. There is no known cure for this condition. Treatment is mainly symptomatic and should be tailored to the patient’s specific needs. In infancy, especially in the neonatal period, maintenance of fluid and electrolyte balance as well as body temperature is of utmost importance. Optimal skin care involves the liberal use of moisturizers/emollients, which may help to repair the epidermal barrier.5 Keratolytic agents such as topical lactic acid, urea, salicylic acid, benzoic acid, or a combination thereof may be used in older children for the treatment of scaly lesions. Mild dandruff shampoos may be used for the treatment of scaling of the scalp. Oral antihistamines may be used to relieve the pruritus.
Topical corticosteroids and/or calcineurin inhibitors have be used to control the underlying inflammation with varying success.34,39 However, care should be taken with topical administration of corticosteroids and calcineurin inhibitors, since children are at risk for marked systemic absorption and associated toxic effects.38 Treatment modalities such as topical retinoids, narrowband UV-B phototherapy, psoralen and ultraviolet irradiation, and oral retinoids (eg, acitretin, isotretinoin) have been used with varying success.7,40,41 Intravenous immunoglobulin and infliximab (a recombinant humanized anti-tumor necrosis factor α [TNF-α] antibody) are new therapeutic options for persons with severe illness.9,10,12,14 Recurrent infections require the use of topical and/or systemic antibiotics. Bacteriophage therapy may be considered in those with severe, chronic, or recurrent infections.17
This patient was treated with oral acitretin, 0.25 mg/kg, and a topical moisturizer, which led to marked improvement of the skin lesions. Genetic counseling was also offered.
Alexander K. C. Leung, MD, is a clinical professor of pediatrics at the University of Calgary and a pediatric consultant at the Alberta Children’s Hospital in Calgary, Alberta, Canada.
Kin Fon Leong, MD, is a consultant pediatric dermatologist at the Pediatric Institute at the Hospital Kuala Lumpur in Kuala Lumpur, Malaysia.
Joseph M. Lam, MD, is a clinical associate professor in the Department of Pediatrics at the University of British Columbia and an associate member at the Department of Dermatology and Skin Sciences at the University of British Columbia, Vancouver, British Columbia, Canada.
REFERENCES:
- Leung AKC, Barankin B, Leong KF. An 8-year-old child with delayed diagnosis of Netherton syndrome. Case Rep Pediatr. 2018;2018:9434916. doi:10.1155/2018/9434916
- Comèl M. Ichthyosis linearis circumflexa. Dermatologica. 1949;98(3):133-136.
- Netherton EW. A unique case of trichorrhexis nodosa—“bamboo hairs.” AMA Arch Derm. 1958;78(4):483-487.
- Trevino J. Netherton syndrome. Clinical Decision Support: Dermatology. 2nd ed. Decision Support in Medicine. https://www.decisionsupportinmedicine.com/DSM/a/articles/2/A/1248. Accessed May 15, 2018.
- Ashton R, Moledina J, Sivakumar B, Mellerio JE, Martinez AE. Considerations in surgical management of a Buschke-Lowenstein tumor in Netherton syndrome: a case report. Pediatr Dermatol. 2017;34(6):e328-e330.
- Mendiratta V, Yadav P, Chander R, Aggarwal S. Recurrent pustular eruption masquerading as pustular psoriasis in Netherton syndrome. Pediatr Dermatol. 2015;32(1):147-148.
- Nevet MJ, Indelman M, Ben-Ari J, Bergman R. A case of Netherton syndrome with intestinal atresia, a novel SPINK5 mutation, and a fatal course. Int J Dermatol. 2017;56(10):1055-1057.
- Dyer JA. Netherton syndrome. UpToDate. https://www.uptodate.com/contents/netherton-syndrome. Updated April 4, 2018. Accessed May 15, 2018.
- Renner ED, Hartl D, Rylaarsdam S, et al. Comèl-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol. 2009;124(3):536-543.
- Roda Â, Mendonça-Sanches M, Travassos AR, Soares-de-Almeida L, Metze D. Infliximab therapy for Netherton syndrome: a case report. JAAD Case Rep. 2017;3(6):550-552.
- De Felipe I, Vázquez-Doval FJ, Vicente J. Comel-Netherton syndrome: a diagnostic challenge. Br J Dermatol. 1997;137(3):468-469.
- Small AM, Cordoro KM. Netherton syndrome mimicking pustular psoriasis: clinical implications and response to intravenous immunoglobulin. Pediatr Dermatol. 2016;33(3):e222-e233.
- De Niear MA, Gigante J. Desquamating rash in a patient with undiagnosed Netherton syndrome. J Pediatr. 2018;192:262-262.e1.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351(2):289-300.
- Goujon E, Beer F, Fraitag S, Hovnanian A, Vabres P. ‘Matchstick’ eyebrow hairs: a dermoscopic clue to the diagnosis of Netherton syndrome. J Eur Acad Dermatol Venereol. 2010;24(6):740-741.
- Meltem Akkurt Z, Tuncel T, Ayhan E, Uçmak D, Uluca Ü, Uçak H. Rapid and easy diagnosis of Netherton syndrome with dermoscopy. J Cutan Med Surg. 2014;18(4):280-282.
- Zhvania P, Hoyle NS, Nadareishvili L, Nizharadze D, Kutateladze M. Phage therapy in a 16-year-old boy with Netherton syndrome. Front Med (Lausanne). 2017;4:94. doi:10.3389/fmed.2017.00094
- Bittencourt MdJS, Moure ERD, Pies OTC, Mendes AD, Deprá MM, de Mello ALP. Trichoscopy as a diagnostic tool in trichorrhexis invaginata and Netherton syndrome. An Bras Dermatol. 2015;90(1):114-116.
- Mubki T, Rudnicka L, Olszewska M, Shapiro J. Evaluation and diagnosis of the hair loss patient: part I. History and clinical examination. J Am Acad Dermatol. 2014;71(3):415.e1-415.e15.
- Söreide K, Söiland H, Körner H, Haga H, Söreide JA. Acute pancreatitis in a young girl with the Netherton syndrome. J Pediatr Surg. 2005;40(11):e69-e72.
- Galadari I, Al-Kaabi J, Galadari H. Netherton syndrome. Skinmed. 2003;2(6):387-389.
- Yerebakan Ö, Uğuz A, Keser İ, et al. Netherton syndrome associated with idiopathic congenital hemihypertrophy. Pediatr Dermatol. 2002;19(4):345-348.
- Pigg MH, Bygum A, Gånemo A, et al. Spectrum of autosomal recessive congenital ichthyosis in Scandinavia: clinical characteristics and novel and recurrent mutations in 132 patients. Acta Derm Venereol. 2016;96(7):932-937.
- Rodríguez-Pazos L, Ginarte M, Vega A, Toribio J. Autosomal recessive congenital ichthyosis. Actas Dermosifiliogr. 2013;104(4):270-284.
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14(2):116-120.
- Leung AKC, Robson WLM. Psoriasis. Consultant Pediatricians. 2005;4(5):240-241.
- Leung AKC, Barankin B. Seborrheic dermatitis. Int J Pediatr Health Care Adv. 2015;2(1):4-6.
- Leung AKC, Barankin B. Acrodermatitis enteropathica. Consultant Pediatricians. 2016;15(8):406-408.
- Cheng R, Yan M, Ni C, Zhang J, Li M, Yao Z. Report of Chinese family with severe dermatitis, multiple allergies and metabolic wasting syndrome caused by novel homozygous desmoglein-1 gene mutation. J Dermatol. 2016;43(10):1201-1204.
- Dănescu S, Leppert J, Cosgarea R, et al. Compound heterozygosity for dominant and recessive DSG1 mutations in a patient with atypical SAM syndrome (severe dermatitis, multiple allergies, metabolic wasting). J Eur Acad Dermatol Venereol. 2017;31(3):e144-e146.
- DiGiovanna JJ. Ichthyosiform dermatosis: so many discoveries, so little progress. J Am Acad Dermatol. 2004;51(1 suppl):S31-S34.
- Stoll C, Alembik Y, Tchomakov D, et al. Severe hypernatremic dehydration in an infant with Netherton syndrome. Genet Couns. 2001;12(3):237-243.
- Krasagakis K, Ioannidou DJ, Stephanidou M, Manios A, Panayiotides JG, Tosca AD. Early development of multiple epithelial neoplasms in Netherton syndrome. Dermatology. 2003;207(2):182-184.
- van der Voort EAM, Prens EP. Netherton syndrome with multiple non-melanoma skin cancers. Acta Derm Venereol. 2013;93(6):727-728.
- Garty BZ, Nimri R. Hypothyroidism in Netherton syndrome. Pediatr Dermatol. 2008;25(1):134-135.
- Macknet CA, Morkos A, Job L, et al. An infant with Netherton syndrome and persistent pulmonary hypertension requiring extracorporeal membrane oxygenation. Pediatr Dermatol. 2008;25(3):368-372.
- Pohl M, Zimmerhackl LB, Hausser I, et al. Acute bilateral renal vein thrombosis complicating Netherton syndrome. Eur J Pediatr. 1998;157(2):157-160.
- Allen A, Siegfried E, Silverman R, et al. Significant absorption of topical tacrolimus in 3 patients with Netherton syndrome. Arch Dermatol. 2001;137(6):747-750.
- Yan AC, Honig PJ, Ming ME, Weber J, Shah KN. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146(1):57-62.
- Lazaridou E, Apalla Z, Patsatsi A, Trigoni A, Ioannides D. Netherton’s syndrome: successful treatment with isotretinoin. J Eur Acad Dermatol Venereol. 2009;23(2):210-212.
- Maatouk I, Moutran R, Tomb R. Narrowband ultraviolet B phototherapy associated with improvement in Netherton syndrome. Clin Exp Dermatol. 2012;37(4):364-366.