
What is Causing This Man's Pain?
A 41-year-old Honduran male with no significant past medical history was admitted to the internal medicine service with progressively worsening myalgias, migratory polyarthralgias, and fever for 10 days. The patient’s history was complicated by recent diagnosis of an allergy to seafood and strep throat. He denied any recent travel, sick contacts, and insect bites. His review of systems was otherwise negative.

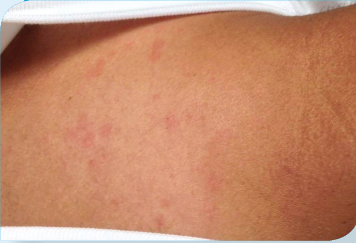
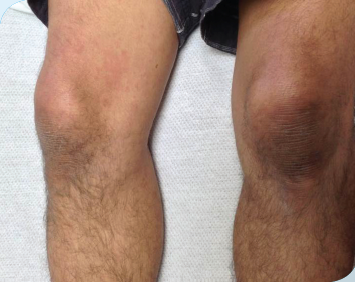
Physical examination. A physical exam demonstrated marked weakness, joint swelling, and tenderness to palpation of several joints and muscle groups. He had 1 swollen and tender posterior cervical lymph node. Over the course of his hospital stay, he developed a diffuse, erythematous rash with both macular and urticarial features that worsened and was mildly pruritic with each episode of fever. These episodes of fever occurred daily, usually in the afternoon or evening, with a maximum temperature of 102.6°F.
Laboratory testing. Basic laboratory tests revealed leukocytosis with a left shift, mildly elevated liver enzymes, elevated erythrocyte sedimentation rate, C-reactive protein, ferritin, and lactate dehydrogenase, but normal antinuclear antibodies, rheumatoid factor, and anticyclic citrullinated peptides. A chest x-ray and CT scan of the abdomen and pelvis were unremarkable.
An extensive infectious work-up was unrevealing, and the patient failed to improve despite the use of several different broad-spectrum antibiotics, including vancomycin, piperacillin/tazobactam, levofloxacin, minocycline, and doxycycline.
Rheumatology and dermatology were consulted. A 4 mm punch biopsy of a lesion on the right arm demonstrated perivascular and interstitial infiltrate with neutrophils and dermal edema.
What's Your Diagnosis? - With Podcast
(Answer and discussion on next page)
Answer: Based on the patient’s clinical picture and laboratory tests, he fulfilled all major, and 4 of 5 minor Yamaguchi criteria, supporting the diagnosis of Adult Onset Still’s Disease.
Discussion
Adult Onset Still’s disease is a rare systemic inflammatory disorder of unknown etiology. It is characterized by spiking fevers, usually exceeding 39°C, that occur at the same time once or twice per day in a quotidian or double quotidian pattern—an evanescent salmon pink rash, arthritis, and multi-organ involvement.1,2
Named after Sir George Frederick Still who first described the symptoms in 22 children; the disorder was named Still’s disease at the time, but is currently known as systemic onset juvenile idiopathic arthritis.3 Adult Onset Still’s Disease was described by Eric Bywaters after noticing 14 adult patients presenting with symptoms similar to the pediatric Still’s disease.4
_________________________________________________________________________________________________________________________________________________________________________________________________________________
RELATED CONTENT
Man With Constant Pain in Left Foot
Vitamin D Deficient Twice as Likely To Have Chronic Pain
________________________________________________________________________________________________________________________________________________________________
The main features of Adult Onset Still’s disease include high spiking fevers, an evanescent rash, polyarthralgias, lymphadenopathy, hepatosplenomegaly, leukocytosis, elevated liver enzymes, erythrocyte sedimentation rate, and ferritin levels.The diagnosis is one of exclusion—as a vast array of infectious etiologies, malignancies, and autoimmune diseases must be ruled out. More commonly encountered differential diagnoses include rheumatic fever, rubella, erythema multiforme, allergic reactions, and meningococcal fever.4
Epidemiology
The exact incidence and prevalence of Adult Onset Still’s Disease is not agreed upon and seems to differ based on region. Cases of Adult Onset Still’s Disease have been reported worldwide. Regardless of the country, the incidence is low, ranging from 0.16 per 100,000 in France and 0.34 per 100,000 in Japan; women are slightly more affected than men.5-7
Presentation
Adult Onset Still’s Disease classically presents with spiking fevers, a rash, and arthralgias or arthritis. Fevers have an estimated incidence of 95.7%, and are described as having a quotidian or double quotidian pattern.1,8,9 Temperatures usually exceed 102.2°F (39°C) and peak in the late afternoon or early evening.1,9 Exacerbation of accompanying symptoms has been noted to coincide with fever spikes.
The rash most associated with Adult Onset Still’s Disease is an evanescent, salmon-pink, maculopapular rash typically found on the proximal limbs and trunk.1,8,9 It is present in close to 73% of patients and may exhibit uncharacteristic features, such as involvement of the face and distal limbs, mild pruritus, and may initially be confused with a drug allergy.1
Arthralgias or arthritis occur in 64% to 100% of cases, with most frequent involvement of the knees, wrists, and ankles, although any joint may be affected.1,8,9 At the onset of disease, joint pain may be migratory, but eventually stabilizes.8 Pharyngitis with a negative physical exam is also common.1,8 These typical symptoms may not all present at the onset of disease, but instead evolve over time.9 Less common disease manifestations include lymphadenopathy, hepatosplenomegaly, myalgias, and/or abdominal pain.8
The natural history of Adult Onset Still’s Disease generally follows 3 distinct patterns with approximately one-third of patients falling into each category.1
- In the monocyclic or self-limited pattern of disease, a single episode of predominately systemic symptoms remits within 1 year.1,8
- With intermittent or polycyclic disease, patients experience a pattern of acute flares and periods of remission.1,8
- The third pattern of disease is chronic and marked by articular involvement that may progress to an erosive arthritis.1,8
Diagnosis
The diagnosis of Adult Onset Still’s Disease is one of exclusion. The differential diagnosis includes many infectious, neoplastic, periodic fever syndromes, and autoimmune diseases. Extensive serologic testing and cultures should be performed to rule out causes, such as connective tissue diseases, systemic lupus erythematosis, vasculitis, viral infections (rubella, parvovirus B19, coxsackie, hepatitis B virus, Ebstein Barr virus, cytomegalovirus, and human immunodeficiency virus), subacute bacterial endocarditis, chronic meningococcemia, tuberculosis, syphilis, and Lyme disease. Important malignancies to rule out include leukemias, lymphomas, and angioblastic lymphadenopathy.
After other etiologies have been ruled out, most rheumatologists use established criteria to diagnose Adult Onset Still’s Disease. The most sensitive criteria used to confirm the diagnosis is the Yamaguchi classification criteria (Table), with a sensitivity of 93.5%.8,9
In the past, Cush’s and Calabro’s criteria were also used to diagnose. However, they are not as sensitive as Yamaguchi’s, both reporting sensitivities of 80.6%.9-11
Treatment
Since the etiology of Adult Onset Still’s Disease is currently unknown, treatment options for these patients are generally targeted at symptom management by decreasing inflammation through the use of non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and disease-modifying anti-rheumatic drugs.
Although NSAIDs are considered to be part of the first line therapy for Adult Onset Still’s Disease, their use alone are only effective in controlling disease in 12% of patients.9 Glucocorticoids, another first line agent, have been shown to be an effective treatment option with most patients requiring at least 1 course during the disease process.1,8,9 Remission should be achieved in most patients with medium-high to high-dose glucocorticoids, with slow tapering over several months to 1 year.8
Methotrexate and other disease-modifying anti-rheumatic drugs have been investigated for the treatment of patients with refractory disease and for decreasing steroid dependence. Although methotrexate was shown to be particularly effective for patients with polyarthritis, this second line agent has not yet been shown to prevent erosive joint damage.8,9
Treatment with biologic agents, such as anakinra (an interleukin-1 receptor antagonist), tocilizumab (an anti-interleukin-6 monoclonal antibody), and anti-tumor necrosis factors, have been reported to show effectiveness as an additional or alternative treatment in some patients.1,8,9
Other proposed therapies include intravenous polyclonal immunoglobulin, cyclosporine, hydroxychloroquine, gold, penicillamine, azathioprine, cyclophosphamide, rituximab, leflunomide, and tacrolimus.1,8,9,14 Of note, canakinumab (Ilaris) is a new monoclonal antibody against interleukin-1 that has recently been approved for the treatment of juvenile inflammatory arthritis, including Still's disease.
More rigorous research, while challenging in rare diseases, is important to further clarify the role of these various agents in the treatment of Adult Onset Still’s Disease.
Outcome of the Case
After ruling out all other etiologies, our patient was put on a course of corticosteroids. Within a few days, his symptoms improved and he was subsequently discharged in stable condition from the hospital. Since then, the patient has followed up with a rheumatologist and is currently undergoing a slow taper of prednisone.
Hear the case discussed in this podcast:
References:
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still’s disease. Ann Rheum Dis. 2006;65:564-572.
- Pouchot J, Vinceneux P. Manifestations cliniques et biologiques de la maladie de Still de l’adulte. Presse Med. 2004;33:1012-1018.
- Still GF. On a form of chronic joint disease in children. Med Chir Trans. 1897;80:47-59.
- Bywaters EG. Still’s disease in the adult. Ann Rheum Dis. 1971;30:121-133.
- Magadur-Joly G, Billaud E, Barrier JH, et al. Epidemiologyof adult Still’s disease: estimate of the incidence by a retrospective study in West France. Ann Rheum Dis. 1995;54:587-590.
- Wakai K, Ohta A, Tamakoshi A et al. Estimated prevalence and incidence of adult Still’s disease: Wndings by a nationwide epidemiological survey in Japan. J Epidemiol. 1997;7:221-225.
- Ohta A, Yamaguchi M, Kaneoka H, et al. Adult Still’s disease: review of 228 cases from the literature. J Rheumatol. 1987;14:1139-1146.
- Fautrel B. Adult-onset Still disease. Best Practice & Research Clinical Rheumatology. 2008;22(5):773-792.
- Bagnari, Colina M, Ciancio G, et al. Adult-onset Still’s disease. Rheumatology International. 2009;30:855-862.
- Yamaguchi M, Ohta A, Tsunematsu T et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424-430.
- Masson C, Le Loet X, Liote F, et al. Comparative study of 6 types of criteria in adult Still’s disease. J Rheumatology. 1996;23:495-497.
- Cush JJ, Medsger TA Jr, Christy WC, et al. Adult onset Still’s disease. Clinical course and outcome. Arthritis Rheum. 1987;30:186-194.
- Calabro JJ, Londino AV Jr. Adult onset Still’s disease. J Rheumatol. 1986;13:827-828.
- Owalia MB, Mehrpoor G. Adult-onset Still’s disease: a review. Indian Journal of Medical Sciences. 2009;63(5):207-221.