
Recognition and Management of Autonomic Dysfunction in Patients With a Lewy Body Disorder: Part II
Lewy body disorders (LBDs) represent a spectrum of neurodegenerative disease, of which Parkinson’s disease (PD) with dementia (PDD) and dementia with Lewy bodies (DLB) are prevalent phenotypes.1 Patients with PD and DLB often develop autonomic dysfunction. Although not a movement disorder like PD nor associated with dementia, pure autonomic failure (PAF) is increasingly recognized as another phenotype of LBD. PAF shares many features with PD and DLB,2 particularly urinary, gastrointestinal, and cardiovascular dysfunction.3,4 Postmortem studies of patients with PAF have observed Lewy bodies in the sympathetic ganglia, but not in the cerebellum, and an accumulation of alfa-synuclein,3 which also occurs in patients with DLB and PD.5 In 2011, Goldstein and associates6 proposed a figure to explain the spectrum and interaction of these disorders.
In part I of this two-part series (http://bit.ly/LBDpart1), we reviewed the proposed origin of cardiovascular autonomic dysfunction in patients with LBD. We also reviewed the symptoms and management of orthostatic hypotension (OH), one of the most significant manifestations of cardiovascular autonomic dysfunction in this patient population. In part II, we examine other important indicators of autonomic failure of the cardiovascular system in patients with LBD, including supine hypertension, postprandial hypotension, carotid sinus syndrome, and rhythm abnormalities. We discuss how to evaluate patients for these cardiovascular phenomena and review management strategies.
Hypertension
Although LBDs are more often associated with hypotension, patients with an LBD may experience any of several types of hypertension (Table 1). The most common are general hypertension, defined as blood pressure ≥140/90 mm Hg; nocturnal hypertension, which is blood pressure ≥120/70 mm Hg during sleep or a failure of mean arterial pressure to decrease by ≥10% during sleep; and supine hypertension, defined as blood pressure ≥140/90 mm Hg when reclining.7 Anecdotal reports suggest that some patients with PAF can even experience exercise-induced hypertension, although this phenomenon appears to be rare.3 Decisions regarding the management of these hypertensive disorders depends on the severity of the condition and the individual patient.

Supine Hypertension
Supine hypertension can be as severe as essential hypertension8 and is increasingly being recognized as a separate clinical disease process in patients with PD and DLB that requires management.7-9 The reported prevalence of supine hypertension among patients with autonomic failure is 50%,7,10 and it is especially prevalent in those with OH.8
When supine hypertension occurs during sleep, it is sometimes referred to as nocturnal hypertension. Whereas supine hypertension can occur any time a patient is laying flat, nocturnal hypertension refers specifically to the failure of mean arterial blood pressure to decrease by ≥10% as part of the normal nocturnal circadian rhythm.11 Nocturnal hypertension is thought to increase the risk of nocturnal natriuresis (sodium loss induced by elevated arterial pressure), contributing to volume loss and a greater likelihood of morning OH.7 Supine hypertension and nocturnal hypertension are typically diagnosed using a 24-hour, ambulatory blood pressure monitor.7
Although the mechanism of supine hypertension in patients with an LBD is not fully understood, it has been linked to dysfunction in the cardiovagal baroreflex (ie, decreased baroreflex gain), which regulates beat-to-beat arterial blood pressure.12 Supine hypertension may also involve pressor mechanisms independent of the sympathetic nervous system.8 Certain medications used to treat OH have been reported to induce supine hypertension in patients and subsequently increase their risk of cardiovascular disease.
The literature is conflicted regarding the impact of supine hypertension on a patient’s health and how to manage it, especially in the setting of concomitant OH.9 The theoretical goal of treating supine hypertension is to decrease blood pressure levels, thus preventing the likelihood of end-organ damage and worsening of OH through pressure natriuresis. Due to the serious risks of prolonged hypertension, however, some have recommended that management of supine hypertension should be aggressive and take precedence over managing OH.7 The counterargument is that failing to treat OH aggressively places patients with an LBD at immediate risk of severe injury (eg, hip and other fractures) due to falls.13
Nonpharmacological therapies should be considered as first-line management for patients with supine hypertension, along with educating patients and any caregivers on the condition, when possible. Options include elevating the head of the bed during sleep or while reclining, decreasing fluid intake before bedtime, and removing compression stockings prior to sleep.
A handful of pharmacological therapies for supine hypertension have been discussed in the literature, such as transdermal nitroglycerine, short-acting calcium channel blockers, and clonidine.7 None of these medications has been studied specifically in the treatment of supine hypertension in patients with PD or DLB and autonomic failure, and their appropriateness as therapy is theoretical. The choice of medication will likely depend on whether the patient is also experiencing OH. When considering pharmacological management of supine hypertension, clinicians may first wish to read some of the in-depth reviews available on this subject.7,8
Postprandial Hypotension
PPH occurs in up to 60% of patients with PD and DLB. This phenomena is defined as a >20 mm Hg drop in systolic blood pressure or a symptomatic decrease in blood pressure that occurs within 2 hours of eating. Like supine hypertension, PPH is frequently associated with OH and can be a severe problem, greatly increasing mortality risk in LBD patients who are >65 years of age.7,14-16 PPH is also thought to worsen parkinsonian symptoms after meals.7,16
PPH may cause syncope or orthostatic intolerance (near syncope) and is thought to result from increased vasodilation in the splanchnic circulation upon consumption of large or carbohydrate-dense meals.13,16,17 Various strategies aimed at preventing vascular dilation have been proposed for managing PPH in this patient population.7,16-18 Nonpharmacological management strategies include eating smaller meals and decreasing overall carbohydrate consumption.
Pharmacological management strategies discussed in the literature include caffeine and octreotide. Caffeine is an adenosine receptor blocker, and it has been suggested that taking 250 mg (the equivalent of approximately 2 cups of coffee) two to three times daily 30 minutes after eating, may reduce the risk of vascular dilation.17,19 Octreotide—a somatostatin analog and a direct vasoconstrictor—has been recommended at a total daily dose of 25 to 200 µg per day, administered subcutaneously every 8 hours.19,20 This drug is extremely expensive, however, and poorly tolerated, with an adverse effects profile that includes nausea, abdominal cramping, and diarrhea.
Carotid Sinus Syndrome
Carotid sinus syndrome (CSS) falls under the umbrella term of neurocardiovascular instability (NCVI), which encompasses OH and vasovagal syndrome. Three variants of carotid sinus syndrome exist (Table 2): cardioinhibitory, diagnosed when cardioinhibition (asystole) lasts ≥3 seconds; vasodepressive, categorized as a ≥50 mm Hg drop in systolic blood pressure during carotid sinus stimulation; and a mixed type that is cardioinhibitory and vasodepressive.21-26 CSS affects nearly 20% of older adults27 and is more prevalent in men.29 In up to 20% of older adults, CSS causes cardiovascular-related syncope21-23,29 and is thought to be an underdiagnosed cause of syncope and falls. Up to 30% of people who test positive for CSS are asymptomatic, however.28
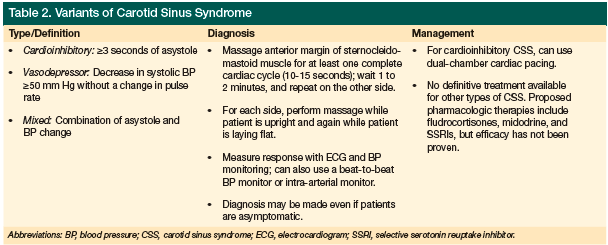
Approximately 40% of patients with DLB and PD are thought to have CSS.21,30 In cases where a CSS-related drop in blood pressure would normally not be sufficient to cause syncope, patients with an LBD are more susceptible to syncope and falling due to the combined effects of their various comorbidities (eg, OH, postural sway/instability, vision problems, and core muscle weakness).
In persons with CSS, carotid sinus stimuli often trigger a bradyarrhythmic response, mediated by the vagus nerve. Even mild neck stimulation can precipitate bradycardia, a sudden drop in blood pressure, and dizziness.28 Response to carotid sinus stimuli is even further exaggerated in patients with DLB.27
Diagnosis of CSS is generally made by massaging the anterior margin of the sternocleidomastoid muscle—which essentially runs from the base of the skull, down and across the side of the neck, to the clavicle—for at least one complete cardiac cycle (approximately 10-15 seconds), waiting 1 to 2 minutes, and repeating the process on the other side. For each side, patients should be evaluated while supine and again in an upright position.31,32 Response is generally measured using a continuous electrocardiogram (ECG) and a blood pressure monitor, but a beat-to-beat blood pressure monitor or intra-arterial monitor can also be used. Before the procedure, patients suspected of having carotid artery disease should undergo ultrasonography to evaluate for critical carotid stenosis.24
Acetylcholinesterase (AChE) inhibitor use may worsen bradyarrhythmic response in patients with CSS and may unmask undiagnosed CSS in patients with PD and DLB.27 Patients with an LBD should therefore be screened for CSS before beginning AChE-inhibitor therapy.24
No therapy has been proven to be effective in managing CSS. Dual-chamber cardiac pacing has been used to treat cardioinhibitory CSS, but has not been found to reduce a patient’s risk of falling.32 The literature includes peripheral discussions of pharmacological therapy for CSS, and a few case reports describe the use of selective serotonin reuptake inhibitor in patients with CSS,33,34 but no one has made any specific recommendations for management.25
Rhythm Abnormalities
QTc prolongation, which may predispose patients to develop arrhythmias, has been observed in patients with PD and DLB. A 2009 study on patients presenting to the emergency department for syncope found that those with PD or DLB were more likely to demonstrate prolongation of the QT interval on ECG.35 Studies involving PD patients have also demonstrated a correlation between the severity of disease and the increased length of the QTc interval.36
Other abnormalities sometimes seen on an ECG in patients with an LBD include decreased heart rate variability with or without bradycardia, which may sometimes be associated with CSS. ECG artifact due to a Parkinson’s tremor may also be evident and sometimes resembles an atrial flutter or polymorphic ventricular tachycardia.37-39
Pacemakers and defibrillators can be used to manage bradycardia and other rhythm abnormalities, and they may improve the patient’s quality of life. Few articles discuss implanting pacemakers and defibrillators in patients with PD or DLB who have deep brain stimulators or implanting deep brain stimulators in patients with pacemakers and defibrillators.40-43 Published case reports, however, suggest that having one device does not necessarily contraindicate implanting the other device.44-46
Conclusion
Cardiac autonomic failure is common in patients with PD, PDD, and DLB, and it can significantly affect quality of life. Part I of this two-part series reviewed OH in patients with an LBD; part II addressed supine hypertension, postprandial hypotension, carotid sinus hypersensitivity, and rhythm abnormalities. Together, this series demonstrates the difficulty clinicians face in managing the symptoms of cardiac autonomic failure in this patient population. These challenges are compounded by the lack of evidence to guide therapeutic decisions.
Furthermore, the cardiovascular system is not the only system affected by Lewy body–induced dysautonomia. When elderly patients present with signs and symptoms of autonomic failure in any bodily system, but especially the cardiovascular system, clinicians should suspect LBD and establish a plan for long-term monitoring.
Until research produces results sufficient to craft evidence-based treatment plans, management of cardiac autonomic failure depends on determining which of the patient’s symptoms have the greatest impact on his or her life and setting small, achievable clinical goals. In addition to improving quality of life, these contributions can help mitigate the risk of falls and their devastating impact on patients with an LBD. The foundation of diagnosis and management of this disease process relies on having open, frank discussion with the patient and his or her caregiver and taking a detailed history of the patient’s symptomatic complaints.
Dr. Walsh recently completed his family medicine internship with Naval Hospital Jacksonville, FL, and is currently a flight surgery student with the Naval Aerospace Medicine Institute in Pensacola, FL. Dr. Unwin is Vice Chair, Department of Family Medicine, Uniformed Services of Health Sciences, Bethesda, MD.
Disclaimer:
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the United States Government.
Copyright Statement:
The authors are military service members. This work was prepared as part of their official duties. Title 17 USC 105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 USC 101 defines a United States Government work as a work prepared by a military service member or employee of the United States Government as part of that person’s official duties.
The authors report no relevant financial relationships.
References
1. Gold G. Dementia with Lewy bodies: clinical diagnosis and therapeutic approach. Front Neurol Neurosci. 2009;24:107-113.
2. Kashihara K, Ohno M, Kawada, Okumura Y. Reduced cardiac uptake and enhanced washout of 123I-MIBG in pure autonomic failure occurs conjointly with Parkinson’s disease and dementia with Lewy bodies. J Nucl Med. 2006;47(7):1099-1101.
3. Sakakibara R, Ogawa E, Kishi M, Uchiyama T, Yamamoto T. Exercise-induced hypertension: A rare manifestation of pure autonomic failure. Eur Neurol J. 2011;2(1):99-101.
4. Horimoto Y, Matsumoto M, Nakazawa H, et al. Cognitive conditions of pathologically confirmed dementia with Lewy bodies and Parkinson’s disease with dementia. J Neurol Sci. 2003;216(1):105-108.
5. Allan LM, Ballard CG, Allen J, et al. Autonomic dysfunction in dementia. J Neurol
Neurosurg Psychiatry. 2007;78(7):671-677.
6. Goldstein DS, Sewell L, Sharabi Y. Autonomic dysfunction in PD: a window to early detection? J Neurol Sci. 2011;310(1-2):118-122.
7. Pathak A, Senard JM. Blood pressure disorders during Parkinson’s disease: epidemiology, pathophysiology and management. Expert Rev Neurother. 2006;6(8):1173-1180.
8. Goldstein DS, Pechnik S, Holmes C, Eldadah B, Sharabi Y. Association between supine hypertension and orthostatic hypotension in autonomic failure [published correction appears in Hypertension. 2003;43(4):e12]. Hypertension. 2002;42(2):136-142.
9. Pathak A, Senard JM. Pharmacology of orthostatic hypotension in Parkinson’s disease: from pathophysiology to management. Expert Rev Cardiovasc Ther. 2004;2(3):393-403.
10. Biaggioni I, Robertson RM. Hypertension in orthostatic hypotension and autonomic dysfunction. Cardiol Clin. 2002;20(2):291-301,vii.
11. Okamoto LE, Gamboa A, Shibao C, et al. Nocturnal blood pressure dipping in the hypertension of autonomic failure. Hypertension. 2009;53(2):363-369.
12. Sharabi Y, Goldstein DS. Mechanisms of orthostatic hypotension and supine hypertension in Parkinson disease. J Neurol Sci. 2011;310(1-2):123-128.
13. Idiaquez J, Roman GC. Autonomic dysfunction in neurodegenerative dementias. J Neurol Sci. 2011;305(1-2):22-27.
14. Fisher AA, Davis MW, Srikusalanukul W, Budge MM. Postprandial hypotension predicts all-cause mortality in older, low-level care residents. J Am Geriatr Soc. 2005;53(8):1313-1320.
15. Senard JM, Chamontin B, Rascol A, Montastruc JL. Ambulatory blood pressure in patients with Parkinson’s disease without and with orthostatic hypotension. Clin Auton Res. 1992;2(2):99-104.
16. Chaudhuri KR, Ellis C, Love-Jones S, et al. Postprandial hypotension and parkinsonian state in Parkinson’s disease. Mov Disord. 1997;12(6):877-884.
17. Sclater A, Alagiakrishnan K. Orthostatic hypotension. A primary care primer for assessment and treatment. Geriatrics. 2004;59(8):22-27.
18. Thomaides T, Karapanayiotides T, Zoukos Y, et al. Gastric emptying after semi-solid food in multiple system atrophy and Parkinson disease. J Neurol. 2005;252(9):1055-1059.
19. Senard JM, Brefel-Courbon C, Rascol O, Montastruc JL. Orthostatic hypotension in patients with Parkinson’s disease: pathophysiology and management. Drugs Aging. 2001;18(7):495-505.
20. Pathak A, Senard JM. Pharmacology of orthostatic hypotension in Parkinson’s disease: from pathophysiology to management. Expert Rev Cardiovasc Ther. 2004;2(3):393-403.
21. Kenny RA, Kalaria R, Ballard C. Neurocardiovascular instability in cognitive impairment and dementia. Ann N Y Acad Sci. 2002;977:183-195.
22. Kapoor JR. Carotid sinus hypersensitivity: a diagnostic pearl. J Am Coll Cardiol. 2009;54(17):1633-1634.
23. Sachpekidis V, Vogiatzis I, Dadous G, Kanonidis I, Papadopoulos C, Sakadamis G. Carotid sinus hypersensitivity is common in patients presenting with hip fracture and unexplained falls. Pacing Clin Electrophysiol. 2009;32(9):1184-1190.
24. Coplan NL. Carotid sinus hypersensitivity and syncope: cause/effect or true/true/unrelated. Arch Intern Med. 2006;166(5):491-492.
25. Freitas J. Carotid sinus syndrome. Management and approach. Rev Port Cardiol. 2004;23(6):903-910.
26. Masson C. Carotid sinus hypersensitivity: an age-related phenomenon. J Neurol Neurosurg Psychiatry. 2006;77(11):1207.
27. Kenny RA, Shaw FE, O’Brien JT, Scheltens PH, Kalaria R, Ballard C. Carotid sinus syndrome is common in dementia with Lewy bodies and correlates with deep white matter lesions. J Neurol Neurosurg Psychiatry. 2004;75(7):966-971.
28. Kerr SR, Pearce MS, Brayne C, Davis RJ, Kenny RA. Carotid sinus hypersensitivity in asymptomatic older persons: implications for diagnosis of syncope and falls. Arch Intern Med. 2006;166(5):515-520.
29. McIntosh SJ, Lawson J, Kenny RA. Clinical characteristics of vasodepressor, cardioinhibitory, and mixed carotid sinus syndrome in the elderly. Am J Med. 1993;95(2):203-208.
30. Wood B, Walker R. Parkinson’s disease: characteristics of fallers and non-fallers. Age Ageing. 2001;30(5):423-424.
31. Parry SW, Richardson DA, O’Shea D, Sen B, Kenny RA. Diagnosis of carotid sinus hypersensitivity in older adults: carotid sinus massage in the upright position is essential. Heart. 2000;83(1):22-23.
32. Parry SW, Steen N, Bexton RS, Tynan M, Kenny RA. Pacing in elderly recurrent fallers with carotid sinus hypersensitivity: a randomised, double-blind, placebo controlled crossover trial. Heart. 2009;95(5):405-409.
33. Dan D, Grubb BP, Mouhaffel AH, Kosinski DJ. Use of serotonin re-uptake inhibitors as primary therapy for carotid sinus hypersensitivity. Pacing Clin Electrophysiol. 1997;20(6):1633-1635.
34. Katz A, Kantor A, Battler A. Serotonin re-uptake inhibitors as primary therapy for carotid sinus hypersensitivity [in Hebrew]. Harefuah. 1998;135(11):505-506, 567.
35. Mussi C, Ungar A, Salvioli G, et al. Evaluation of Guidelines in Syncope Study 2 Group. Orthostatic hypotension as cause of syncope in patients older than 65 years admitted to emergency departments for transient loss of consciousness. J Gerontol A Biol Sci Med Sci. 2009;64(7):801-806.
36. Oka H, Mochio S, Sato H, Katayama K. Prolongation of QTc interval in patients with Parkinson’s disease. Eur Neurol. 1997;37(3):186-189.
37. Vanerio G. Tremor as a cause of pseudoatrial flutter. Am J Geriatr Cardiol. 2007;16(2):
106-108.
38. Bhatia L, Turner DR. Parkinson’s tremor mimicking ventricular tachycardia. Age Ageing. 2005;34(4):410-411.
39. Boos CJ, Khan MY, Thorne S. An unusual case of misdiagnosed ventricular tachycardia. Emerg Med J. 2008;25(3):173-174.
40. Ozben B, Bilge AK, Yilmaz E, Adalet K. Implantation of a permanent pacemaker in a patient with severe Parkinson’s disease and a preexisting bilateral deep brain stimulator. Int Heart J. 2006;47(5):803-810.
41. Capelle HH, Simpson RK Jr, Kronenbuerger M, Michaelsen J, Tronnier V, Krauss JK. Long-term deep brain stimulation in elderly patients with cardiac pacemakers. J Neurosurg. 2005;102(1):53-59.
42. Obwegeser AA, Uitti RJ, Turk MF, et al. Simultaneous thalamic deep brain stimulation and implantable cardioverter-defibrillator. Mayo Clin Proc. 2001;76(1):87-89.
43. Senatus PB, McClelland S 3rd, Ferris AD, Ford B, Winfield LM, Pullman SL. Implantation of bilateral deep brain stimulators in patients with Parkinson disease and preexisting cardiac pacemakers. Report of two cases. J Neurosurg. 2004;101(6):1073-1077.
44. Ozben B, Bilge AK, Yilmaz E, Adalet K. Implantation of a permanent pacemaker in a patient with severe Parkinson’s disease and a preexisting bilateral deep brain stimulator. Int Heart J. 2006;47(5):803-810.
45. Capelle HH, Simpson RK Jr, Kronenbuerger M, Michaelsen J, Tronnier V, Krauss JK. Long-term deep brain stimulation in elderly patients with cardiac pacemakers. J Neurosurg. 2005;102(1):53-59.
46. Obwegeser AA, Uitti RJ, Turk MF, et al. Simultaneous thalamic deep brain stimulation and implantable cardioverter-defibrillator. Mayo Clin Proc. 2001;76(1):87-89.