
Pharmacological Therapy of Alzheimer’s Disease: Current and Investigated Treatments
Abstract: Alzheimer’s disease affects 5.3 million persons in the United States and is the fifth leading cause of death of Americans 65 years and older. Although there is no cure for Alzheimer’s disease, there is a growing understanding of its pathophysiology, which is helping researchers to develop more effective therapies that may halt or even reverse the pathogenesis of this disease. In addition to reviewing current and experimental treatment options for patients with Alzheimer’s disease, the author discusses clinical studies investigating whether various pharmacologic agents indicated for other illnesses might benefit patients with this dementia.
_________________________________________________________________________________________________________
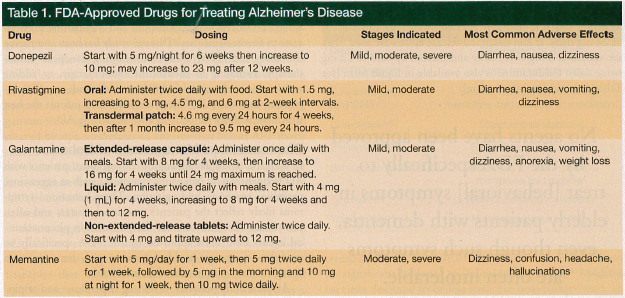
Alzheimer’s disease (AD), which causes cognitive deterioration and death, affects 5.3 million persons in the United States and is the fifth leading cause of death among Americans 65 years and older.1 Nearly half of Americans with AD have moderate to severe disease.2 In 2010, the United States spent an estimated $172 billion to care for people 65 years and older with AD and other dementias; this figure does not include the value of care provided by unpaid caregivers, which was estimated at $144 billion in 2009.1 The pharmacologic treatments available for AD (Table 1), none of which are curative, have limited efficacy at addressing symptoms. Better therapies—especially ones that halt or even reverse pathogenesis of the disease—are desired.
Although we do not yet fully understand what causes AD or how it progresses, a steady stream of data continues to shed light on the disease’s pathophysiology. This growing understanding has spurred efforts to develop more effective therapies, including vaccines and monoclonal antibodies. In addition to reviewing current and experimental treatment options, we discuss clinical studies investigating whether various pharmacologic agents indicated for other illnesses might benefit patients with AD.
Cholinesterase Inhibitors
For decades, researchers have known that AD involves a loss of cholinergic input to the cortex from the basal forebrain. A classic 1982 study by Whitehouse and associates3 reported dramatic depletion of cholinergic neurons in the nucleus basalis of Meynert in patients with AD. Cholinergic neurons express choline acetyltransferase, an enzyme that synthesizes the neurotransmitter acetylcholine (ACh), which is essential for several nervous system functions, including memory.2 Subsequent studies involving histology, positron emission tomography, and magnetic resonance imaging (MRI) support the loss of cholinergic function as a major feature of AD and suggest a parallel between decreasing cholinergic activity and increasing disease severity.2 Evidence of a relationship between cholinergic deficits and AD led to the development of drugs designed to inhibit acetylcholinesterase (AChE), an enzyme that metabolizes ACh at the cholinergic synapses.2
Although inhibiting AChE prolongs synaptic cholinergic activity, cholinesterase inhibitors are not disease-modifying drugs, meaning they do not affect the fundamental pathophysiology of AD that is thought to cause neurodegeneration, abnormalities of beta-amyloid (Abeta), and hyperphosphorylation of tau proteins. By improving cholinergic transmission, AChE inhibitors may promote apparent clinical stability or even improvement in the early (mild to moderate) stages of the disease, but there is no convincing evidence that they affect the disease course or prognosis. As the underlying disease continues to progress, more cholinergic neurons are lost, compromising the efficacy of AChE inhibitors.
Guidelines from the Quality Standards subcommittee of the American Academy of Neurology recommend AChE inhibitors as standard therapy (a category for which there is the highest degree of clinical evidence from randomized controlled studies) for AD.4 AChE inhibitors can produce increased extracerebral acetylcholinergic activity, and adverse effects associated with their use include diarrhea, nausea, bradycardia, and syncope.
Tacrine was the first AChE inhibitor approved by the US Food and Drug Administration (FDA) for AD, but due to its very short half-life and hepatotoxicity, it has since gone out of use. The three AChE inhibitors currently used in clinical practice to treat patients with mild to moderate AD are donepezil, rivastigmine, and galantamine. Donepezil has also been approved by the FDA for use in severe AD. Studies comparing these three cholinesterase inhibitors have not consistently shown one to be superior to another.5
Although numerous studies of drugs in this class have demonstrated statistically significant improvement in cognition measures for patients with AD, the level of clinical improvement is usually small.6 A patient’s clinical status may reflect a balance between the drug’s ability to promote cholinergic activity of the surviving neurons and ongoing neurodegeneration, complicating efforts to quantify the level of benefit a patient receives from the drug. It is possible AChE inhibitors may only slow the rate of decline, but even this can be worthwhile to patients and their caregivers. Research continues into drugs that enhance cholinergic transmission, including nicotinic receptor agonists.7
Donepezil
Of the three currently used AChE inhibitors, donepezil is the earliest to be approved and is effective in treating patients in the mild, moderate, and severe stages of AD.8 The starting dose of donepezil is 5 mg daily. After 6 weeks, the dose is increased to 10 mg daily if tolerated. Evidence suggests donepezil dosed at 10 mg per day is less likely to cause adverse effects than comparable doses of the other AChE inhibitors.5
A generic formulation of donepezil 10 mg became available in 2009. Shortly thereafter, the drug’s original manufacturer released a 23-mg pill available only under their brand name for daily dosing; this dose was said to be more effective than the 10-mg daily dose.2 The FDA approved the higher-dose formulation based on data from a randomized, double-blind, multicenter, international study that appeared to demonstrate its superior efficacy against the 10-mg daily dose on two predetermined measures.9 The first measure is the Severe Impairment Battery (SIB) instrument, which assesses memory; language; orientation; attention; praxis; and visuospatial, constructional, and social abilities. The other is the Clinician’s Interview-Based Impression of Change incorporating caregiver information (CIBIC-Plus), which uses caregiver input in rating the patient’s condition from 1 (markedly improved) to 7 (markedly worse). Overall, the 23-mg daily dose demonstrated a statistically significant benefit compared with the 10-mg daily dose when outcomes were measured using the SIB but not with the CIBIC-Plus. Although the improvement seen with the SIB was statistically significant, it was also small and might not be perceptible to the patient or caregiver, leading some to question the value of the 23-mg daily dose.
An analysis of data for the subset of patients with more severe AD—which was not part of the original study design—showed statistically significant gains on the SIB and on the CIBIC-Plus with the 23-mg daily dose; however, it is debatable whether the post-hoc identification of subgroups for analysis produces valid results, and approval of the 23-mg dose of donepezil remains the subject of controversy. A group called Public Citizen has petitioned the FDA to rescind approval of the 23-mg formulation, expressing concerns about its safety and questioning whether data are sufficient to support the claims of its superior efficacy.10
Rivastigmine
Rivastigmine, which has a shorter half-life than donepezil, inhibits both central AChE and butyrylcholinesterase, but it is not clear whether this translates to increased clinical efficacy. Rivastigmine is available in oral and transdermal formulations, which have been approved by the FDA for use in patients with AD- or Parkinson’s-related dementia. The starting dose of the oral preparation is 1.5 mg twice daily, which is gradually titrated upward to 6 mg twice daily. The starting dose for the transdermal formulation is 4.6 mg daily, which can be increased to 9.5 mg daily after 1 month. A recent double-blind, randomized study compared rivastigmine capsules with the patch in patients with probable AD and found both improved activities of daily living.11
Of the three cholinesterase inhibitors available, oral rivastigmine has the highest incidence of adverse effects (eg, nausea, vomiting, and dizziness).5 In addition to producing a lower rate of gastrointestinal side effects, the transdermal rivastigmine patch is easier to use for patients resistant to taking oral medications.12
Galantamine
Galantamine, like rivastigmine, has a shorter half-life than donepezil. Galantamine is available in an extended-release formulation that is administered once daily at a starting dose of 8 mg, which is gradually titrated upward to a maximum of 24 mg daily. If non-extended-release tablets are used, they are started at 4 mg twice daily and titrated upward to 12 mg twice daily. Galantamine is also available in liquid form (see Table 1 for dosing information).
Galantamine was initially approved in 2001 to treat mild to moderate AD based on clinical trial data that showed 16-mg and 24-mg daily doses of galantamine produced statistically significant improvement in AD Assessment Scale–Cognitive Subscale Scores and CIBIC-Plus outcomes compared with placebo.13 In controlled clinical trials, the most frequently reported adverse events associated with galantamine included nausea, vomiting, diarrhea, anorexia, and weight loss.
Memantine
Memantine is an uncompetitive inhibitor of N-methyl-d-aspartate (NMDA) receptor–mediated glutamate channels. It binds only to channels that are already open, thereby helping to reduce excessive excitation of neurons via such channels, which can cause neuronal dysfunction and damage. NMDA-mediated channels bind the neurotransmitter glutamate and play a role in normal learning and memory.
Because memantine only blocks excessive activity, rather than all glutamate binding, it should not interfere with normal neuronal activity. Memantine has a long half-life of 60 to 80 hours. The starting dose is 5 mg daily, which is gradually titrated upward to 10 mg twice daily. Side effects associated with memantine include dizziness, headache, confusion, and hallucinations, despite the theoretical advantages of uncompetitive binding.
Memantine has been found to modestly improve cognitive function in patients with moderate to severe AD.14-16 A recent meta-analysis of studies investigating memantine found it was no better than placebo, however, in treating patients with mild (defined as a Mini-Mental State Examination [MMSE] score of 20-23) to moderate (MMSE score of 10-19) AD.17 There is evidence that combination therapy with memantine and donepezil is beneficial.18 The ongoing randomized, controlled DOMINO-AD (Donepezil and Memantine in Moderate to Severe Alzheimer’s Disease) study has been comparing the safety and effectiveness of continuing donepezil monotherapy, switching to memantine monotherapy, or adding memantine to donepezil for patients with moderate AD progressing to severe AD.19 Results should help inform the best treatment strategy for patients with advancing AD.
Pharmaceutical Management of Behavior
In addition to displaying cognitive symptoms, patients with AD frequently have behavioral symptoms such as aggression, paranoia, psychosis, and agitation. These behavioral symptoms likely reflect the patient’s emotional distress and often complicate efforts to care for the patient. No pharmaceutical agents have been approved by the FDA specifically to treat these symptoms in elderly patients with dementia, even though such symptoms are often intolerable.
Atypical antipsychotics, including risperidone and aripiprazole, have demonstrated modest benefit at ameliorating these behavioral symptoms when used for up to 12 weeks.20 A Cochrane review of atypical antipsychotic use in patients with dementia found evidence that risperidone and olanzapine reduce aggression and that risperidone reduces psychosis, but both drugs were associated with increases in stroke risk and extrapyramidal symptoms.21 Other investigations of typical and atypical antipsychotic treatment in elderly persons with dementia have reported an increased rate of mortality.22 The evidence suggests the benefits of antipsychotic treatments are too small to justify the risks.23
Elderly patients with dementia may tolerate antidepressants, such as citalopram, sertraline, or trazodone, better than antipsychotics, but few data support their effectiveness at treating behavioral problems in this patient population.24 However, because antidepressants appear to be a safer option than antipsychotics for dementia patients, it is reasonable to try antidepressants first when attempting to treat behavioral symptoms pharmacologically.
Investigational Disease-Modifying Drugs
 Concerted effort is under way to develop drugs that modify the course of AD, and several trials of investigational therapies in patients with AD and dementia are ongoing. Many novel therapies have focused on addressing Abeta peptide and hyperphosphorylated tau protein abnormalities, which play a major role in the pathophysiology of AD.25 Additional approaches include new or existing compounds that could repair or prevent some of the negative effects of Abeta or agents that interact with other possibly relevant targets. Many major, randomized, placebo-controlled studies have been conducted in pursuit of new, more effective pharmacological therapies for AD, but little success has been reported (Table 2).26-33
Concerted effort is under way to develop drugs that modify the course of AD, and several trials of investigational therapies in patients with AD and dementia are ongoing. Many novel therapies have focused on addressing Abeta peptide and hyperphosphorylated tau protein abnormalities, which play a major role in the pathophysiology of AD.25 Additional approaches include new or existing compounds that could repair or prevent some of the negative effects of Abeta or agents that interact with other possibly relevant targets. Many major, randomized, placebo-controlled studies have been conducted in pursuit of new, more effective pharmacological therapies for AD, but little success has been reported (Table 2).26-33
Abeta-Targeting Therapies
Abeta, particularly its 42-amino acid isoform (Abeta42), aggregates in extracellular plaques. These plaques feature prominently in the histological analysis of brain tissue specimens from deceased patients with AD. Abeta is generated when amyloid precursor protein (APP), a normal transmembrane protein, is sequentially cleaved by the beta-secretase and gamma-secretase enzymes. Abeta does not result when APP is cleaved by alpha-secretase, an enzyme that breaks APP in the middle of a segment essential for Abeta production. Based on these findings, drugs that prevent gamma-secretase or beta-secretase from cleaving APP or that promote cleavage of APP by alpha-secretase might be useful as disease-modifying agents for AD.
Abeta aggregates in the brain, forming oligomers and plaques, and evidence suggests it is the soluble Abeta oligomers that are responsible for damaging neurons.34 Novel therapies that interfere with Abeta aggregation or target gamma-secretase and thus decrease Abeta production have been investigated, in some cases progressing to phase 3 clinical trials, but results have been disappointing.7,33,35 Tarenflurbil32 and semagacestat30 are experimental agents that act on gamma-secretase, whereas tramiprosate is an Abeta antiaggregant.33
Because gamma-secretase has important functions besides APP processing, such as processing the Notch protein, researchers have looked at biochemical pathways for blocking APP processing that do not inhibit Notch processing.36 Beta-secretase is one possible target, but efforts to target this enzyme have been challenging due to the difficulty getting the bulky inhibiting compounds across the blood–brain barrier.35,37 One study harnessed exosomes—tiny vesicles that cells manufacture for transporting protein or RNA—to deliver compounds of small interfering RNA specific for beta-secretase to the brain cells of mice, greatly reducing beta-secretase activity.38 If this approach proved feasible for human use, it could help resolve the problem of getting beta-secretase inhibitors across the blood–brain barrier. Clinically, upregulation of alpha-secretase remains a more remote target.7,35
Results of studies into the mechanics of AD suggest clearing Abeta from the brain may represent another direction for AD drug research. It is well known that the ε4 allele of the apolipoprotein E (APOE) gene is a major genetic risk factor for AD. Castellano and colleagues39 recently discovered that APOE plays an important role in regulating clearance of Abeta from the brain and that presence of the ε4 allele inhibits Abeta clearance. Immunotherapy with targeted Abeta vaccines is also being investigated as an approach for reducing Abeta burden. Based on the effectiveness of an immunotherapy vaccine specific for Abeta42 in reducing plaque in the brains of murine AD models, investigators initiated a clinical trial in humans. The trial was terminated early because of safety concerns and produced no significant evidence of improved cognition.26 Modified vaccines designed to be less toxic are now being tested, including ACC-001, which has already advanced to clinical trials.37 With data from one early-stage Abeta42 immunotherapy trial demonstrating that clearance of Abeta plaque did not correspond with prevention of end-stage dementia and death,40 it remains to be seen whether Abeta vaccines will be clinically effective.
Bapineuzumab, a humanized monoclonal antibody designed to clear Abeta, is being investigated in ongoing trials. The efficacy of bapineuzumab has not yet been established; preliminary data show some patients taking the drug developed vasogenic edema and some demonstrated possible cognitive improvement.41
A recent hypothesis for why drugs that act on Abeta production or clearance fail to treat AD suggests that Abeta exerts its pathogenic effects early in the disease process, perhaps even before individuals demonstrate symptoms, and triggers a cascade of neuron-, glial-, and synapse-damaging effects that over time cause dementia symptoms. It has been theorized that activation of the enzymes calcineurin and caspases, which play a major role in these cascades, progress independently of Abeta.42 If this theory is correct, it might be necessary to administer drugs that act on Abeta early in the disease process. Doing so, however, would likely require identifying biomarkers of preclinical disease, an area of active research that is beyond the scope of this review.
Tau-Targeted Approaches
Intracellular tangles are another prominent histological hallmark of AD. These tangles comprise paired helical filaments made up of hyperphosphorylated tau protein. Studies have been investigating the effectiveness of methylene blue (methylthioninium chloride) at inhibiting tau aggregation. Preliminary data from a phase 2 trial of methylene blue in patients who were not taking AChE inhibitors or memantine suggested patients taking methylene blue experienced significant cognitive improvement compared with placebo, but it does not appear that a full report of the findings has been published. Because methylene blue turns urine blue, the drug cannot be investigated in a blinded fashion, which could have an effect on outcomes.The manufacturer of methylene blue says it is working on a modified form of the drug (LMTX) that the company
anticipates will be better tolerated.43
Another approach is to block one of the many kinases that hyperphosphorylate tau. Glycogen synthase kinase is one target, and data show lithium and depakote inhibit this kinase. To date, no study has demonstrated any benefit in AD with use of lithium or other kinase inhibitors, but various kinase-inhibiting drugs are under development. Two kinase inhibitors being tested in clinical trials are davunetide, which appears to have some inhibitory effects on tau phosphorylation, and nicotinamide, which decreases some hyperphosphorylated forms of tau.7
Other Approaches
Abeta may damage mitochondria, and impaired mitrochondrial function has been associated with oxidative injury to cells. Latrepirdine is an antihistamine that has a number of physiological effects, of which its ability to stabilize mitochondria may be the most relevant for patients with AD. A Russian trial investigating latrepirdine in patients with mild to moderate AD reported promising results in 2008.28 A more recent multinational study was terminated, however, after preliminary data showed no benefit.29
Zinc and copper may affect Abeta aggregation and modulate NMDA-receptor activity.7,33 The drugs clioquinol and PBT2 alter the interaction of these metals with Abeta. Although PBT2 is similar to clioquinol, it is less toxic and has been the focus of preliminary studies.7,33
APOE is the major cholesterol transporter in the brain, and cholesterol plays a role in the cleavage of APP into Abeta, which occurs in specific membrane regions called lipid rafts. The possible role of cholesterol in the pathophysiology of AD has prompted studies into the effectiveness of statins against AD; however, a recent large, randomized, controlled clinical trial that treated patients with AD with simvastatin, a drug that crosses the blood–brain barrier, found no improvement in cognition.31
Advanced glycation end-products (AGEs) may help Abeta transverse the blood–brain barrier and
enter the cerebrospinal fluid, and oxidative damage may result from binding of AGE to its receptor.7 AGE molecules are formed when sugar residues react with protein amino groups. AGEs promote cross-linking of proteins and continue to accumulate as part of the aging process. In a pilot study, an AGE receptor inhibitor showed safety but not efficacy in treating Alzheimer’s disease.44
Other connections between abnormal glucose processing and AD are being studied, including the relationship of insulin, a hormone involved in multiple brain functions.45 In addition to insulin’s effects on glucose metabolism, insulin may mitigate pathological binding of Abeta oligomers to hippocampal synapses and reduce glycogen synthase kinase hyperphosphorylation of tau. Studies are exploring intranasal insulin—hypothesized to deliver insulin directly into the brain—as a treatment for AD, with a recently published study showing some promising results.46,47
Peroxisome proliferator-activated receptor (PPAR) gamma is a transcription factor that, when stimulated, may reduce beta-secretase activity.37 PPARs are also involved in metabolism of glucose and lipids and in inflammation. PPAR-gamma agonists are currently approved to treat diabetes mellitus. A meta-analysis that included three pilot trials and nine clinical trials involving the PPAR-gamma agonists pioglitazone or rosiglitazone (which does not cross the blood–brain barrier well) in patients with AD or mild cognitive impairment reported variable results and no definitive evidence supporting their effectiveness.45 Given the growing concern regarding the cardiac effects of these drugs, enthusiasm even for their use in diabetes has waned.
Studies have established a correlation between eating fish, which is high in docosahexaenoic acid (DHA), and a reduced risk of dementia and AD. This prompted a large, randomized, placebo-controlled trial of DHA supplementation in individuals with mild to moderate AD.27 Patients given DHA supplements failed to demonstrate any cognitive gains or improvement in brain atrophy, as measured using MRI, compared with patients given a placebo.27
Conclusion
Currently approved pharmacological treatments for AD consist of central cholinesterase inhibitors and memantine, whose benefits are modest but well documented. Clinical trials for novel disease-modifying drugs or new approaches have thus far produced disappointing results. Researchers continue to pursue drugs designed to reduce Abeta generation, block toxicity of Abeta oligomers, increase Abeta clearance from the brain, and block hyperphosphorylation of tau. Evidence indicating an earlier amyloid-dependent phase and a subsequent amyloid-independent phase in the pathophysiology of AD suggests timing of treatment may be crucial. Limited evidence supports pharmacologic treatment of AD behavioral symptoms. If considered, antidepressants may be a more appropriate initial therapeutic option, given the increased mortality demonstrated with the use of antipsychotic drugs in clinical trials.
The author reports no relevant financial relationships.
References
1. Alzheimer’s Association. 2010 Alzheimer’s disease facts and figures. Alzheimers Dement. 2010;6(2):158-194.
2. Sabbagh M, Cummings J. Progressive cholinergic decline in Alzheimer’s disease: consideration for treatment with donepezil 23 mg in patients with moderate to severe symptomatology. BMC Neurol. 2011;11:21.
3. Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215(4537):1237-1239.
4. Doody RS, Stevens JC, Beck RN, et al. Practice parameter: management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56(9):1154-1166.
5. Hansen RA, Gartlehner G, Webb AP, Morgan LC, Moore CG, Jonas DE. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: a systematic review and meta-analysis. Clin Interv Aging. 2008;3(2):211-225.
6. Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008;148(5):379-397.
7. Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer’s disease: clinical trials and drug development [published correction appears in Lancet Neurol. 2010;10(6):501]. Lancet Neurol. 2010;9(7):702-716.
8. Cummings J, Jones R, Wilkinson D, et al. Effect of donepezil on cognition in severe Alzheimer’s disease: a pooled data analysis. J Alzheimers Dis. 2010;21(3):843-851.
9. Farlow MR, Salloway S, Tariot PN, et al. Effectiveness and tolerability of high-dose (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: a 24-week, randomized, double-blind study. Clin Ther. 2010;32(7):1234-1251.
10. Valeo T. Medical watchdog group asks FDA to withdraw donepezil 23 mg. Neurology Today. 2011;11(14):1,9,10.
11. Grossberg G, Meng X, Olin JT. Impact of rivastigmine patch and capsules on activities of daily living in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2011;26(1):65-71.
12. Small G, Bullock R. Defining optimal treatment with cholinesterase inhibitors in
Alzheimer’s disease. Alzheimers Dement. 2011;7(2):177-184.
13. Tariot PN, Solomon PR, Morris JC, et al; the Galantamine USA-10 Study Group. A 5-month, randomized, placebo-controlled trial of galantamine in AD. Neurology. 2000;54(12):2269-2276.
14. Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ; Memantine Study Group. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333-1341.
15. Areosa SA, Sheriff F, McShane R. Memantine for dementia. Cochrane Database Syst Rev. 2005;3:CD003154.
16. Herrmann N, Li A, Lanctôt K. Memantine in dementia: a review of the current evidence. Expert Opin Pharmacother. 2011;12(5):787-800.
17. Schneider LS, Dagerman KS, Higgins JPT, McShane R. Lack of evidence for the efficacy of memantine in mild Alzheimer’s disease. Arch Neurol. 2011;68(8):991-998.
18. Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I; Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
19. Jones R, Sheehan B, Phillips P, et al; DOMINO-AD team. DOMINO-AD protocol: donepezil and memantine in moderate to severe Alzheimer’s disease—a multicentre RCT. Trials. 2009;10:57.
20. Ballard C, Creese B, Corbett A, Aarsland D. Atypical antipsychotics for the treatment of behavioral and psychological symptoms in dementia, with a particular focus on longer term outcomes and mortality. Expert Opin Drug Saf. 2011;10(1):35-43.
21. Ballard C, Waite J. The effectiveness of atypical antipsychotics for the treatment of aggression and psychosis in Alzheimer’s disease. Cochrane Database Syst Rev. 2006;25(1):CD003476.
22. Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353(22):2335-2341.
23. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525-1538.
24. Seitz DP, Adunuri N, Gill SS, Gruneir A, Herrmann N, Rochon P. Antidepressants for agitation and psychosis in dementia. Cochrane Database Syst Rev. 2011;2:CD008191.
25. Querfurth HW, LaFerla FM. Alzheimer’s disease [published correction appears in
N Engl J Med. 2011;364(6):588]. N Engl J Med. 2010;362(4):329-344.
26. Gilman S, Koller M, Black RS, et al; AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553-1562.
27. Quinn JF, Raman R, Thomas RG, et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer’s disease: a randomized trial. JAMA. 2010;304(17):1903-1911.
28. Doody RS, Gavrilova SI, Sano M, et al; Dimebon Investigators. Effects of dimebon on cognition, activities of daily living, behavior, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled study. Lancet. 2008;372(9634):207-215.
29. Miller G. Pharmacology. The puzzling rise and fall of a dark-horse Alzheimer’s drug. Science. 2010;327(5971):1309.
30. Samson K. NerveCenter: phase III Alzheimer trial halted: search for therapeutic biomarkers continues. Ann Neurol. 2010;68(4):A9-A12.
31. Sano M, Bell KL, Galasko D, et al. A randomized, double-blind placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2011;77(6):556-563.
32. Green RC, Schneider LS, Amato DA, et al; Tarenflurbil Phase 3 Study Group. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302(23):2557-2564.
33. Galimberti D, Scarpini E. Disease-modifying treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2011;4(4):203-216.
34. Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837-842.
35. Carter MD, Simms GA, Weaver DF. The development of new therapeutics for Alzheimer’s disease. Clin Pharmacol Ther. 2010;88(4):475-486.
36. Gandy S, Wustman B. New pathway links gamma-secretase to inflammation and memory while sparing notch. Ann Neurol. 2011;69(1):5-7.
37. Chopra K, Misra S, Kuhad A. Current perspectives on pharmacotherapy of Alzheimer’s disease. Expert Opin Pharmacother. 2011;12(3):335-350.
38. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29(4):341-345.
39. Castellano JM, Kim J, Holtzman DM, et al. Human apoE isoforms differentially regulate brain amyloid-ß peptide clearance. Sci Transl Med. 2011;3(89):89ra57.
40. Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216-223.
41. Salloway S, Sperling R, Gilman S, et al; Bapineuzumab 201 Clinical Trial Investigators. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer’s disease. Neurology. 2009;73(24):2061-2070.
42. Hyman BT. Amyloid-dependent and amyloid independent stages of Alzheimer disease. Arch Neurol. 2011;68(8):1062-1064.
43. TauRX Pharmaceuticals. Tau aggregation inhibitors as therapeutic agents. www.taurx.com/tau.htm. Accessed February 27, 2012.
44. Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25(3):206-212.
45. Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment? Br J Clin Pharmacol. 2011;71(3):365-376.
46. Benedict C, Frey WH 2nd, Schiöth HB, Schultes B, Born J, Hallschmid M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp Gerontol. 2011;46(2-3):112-115.
47. Craft S, Baker LD, Montine TJ, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69(1):29-38.