
Pfeiffer Syndrome Type 2: Cloverleaf-Shaped Skull in a Newborn
A girl was born at 37 weeks of gestation to a 40-year-old gravida 3, para 2 mother via cesarean delivery at a county hospital. The infant had an Apgar score of 8 at 1 minute and 9 at 5 minutes. The infant was stable on room air at delivery but was transferred to the neonatal intensive care unit (NICU) for further monitoring.
The pregnancy had been uncomplicated, and maternal laboratory test results were negative for HIV, group B streptococci, syphilis, and rubella. At 14 weeks, fetal ultrasonography showed no visible abnormalities; however, magnetic resonance imaging (MRI) of the mother’s abdomen showed a cloverleaf deformity of the fetus’s skull, syndactyly of the left middle finger and ring finger, hypertelorism of the orbits with proptosis of the eyes, and low-set ears.
The mother’s medical history is not significant, and there is no history of consanguinity or congenital disorders in her family or in the father’s family. She has two other healthy children with the same father.

Physical Examination
The infant’s vital signs had been relatively normal at birth and throughout the NICU stay. Birth weight, length, and head circumference were in the normal range for gestational age.
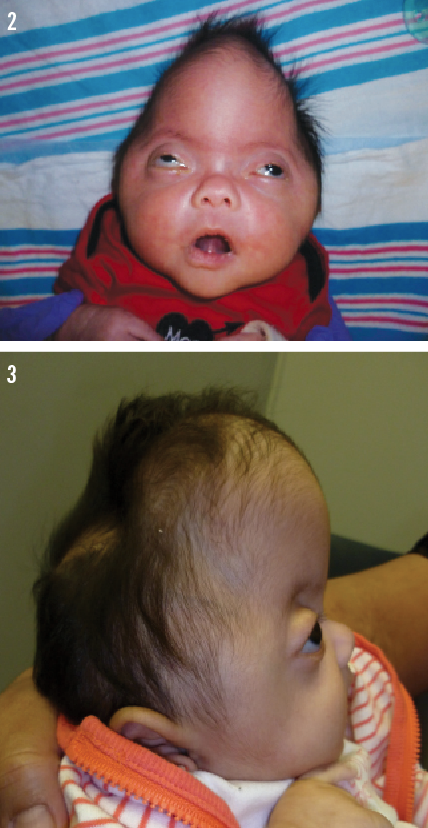
Physical examination showed a closed sagittal suture, a laterally fused coronal suture, and open temporal, metopic, and lambdoid sutures (Figures 1-3). Midfacial hypoplasia was noted, along with a flat nasal bridge and a small mandible. Bilateral exophthalmos and low-set ears were present. A catheter was passed through both nares with moderate difficulty. Breath sounds were present; however, noisy upper airway sounds were heard. The infant had good range of motion of the extremities, including at the elbows. Syndactyly of the second and third digits of the hands and feet was present (Figures 4 and 5). She had short proximal and middle phalanges and broad distal phalanges.
The rest of the physical examination findings were unremarkable. Heart rate was regular without murmur, and bowel sounds were present. The abdomen was soft, with no organomegaly.
Based on the physical examination findings, the infant received a diagnosis of Pfeiffer syndrome type 2.
Further Testing
A radiographic skeletal survey showed the characteristic features of Pfeiffer syndrome, including the cloverleaf skull deformity (Figure 6); small, shallow orbits; broad great toes with deformed metatarsals and phalanges (Figure 7); a supernumerary digit on the left hand; partial syndactyly of the bones and index fingers of both hands; and possible left radioulnar synostosis.
A maxillofacial computed tomography scan (Figure 8) showed multiple bony abnormalities and ruled out choanal atresia. Out of concern about visceral abnormalities possible with Pfeiffer syndrome, chest radiography and echocardiography was done. Results of chest radiography were normal; echocardiography showed a small midmuscular ventricular septal defect with left-to-right shunt, and a small patent foramen ovale with left-to-right shunt.
An MRI scan of the brain and spine showed brachycephaly with marked deformity of the frontal calvaria and upward displacement of the frontal lobes. No hydrocephaly or intracranial hemorrhage was seen.
An upper gastrointestinal series showed normal gastric emptying and no structural abnormalities. No genetic testing was done for this patient.

Pfeiffer Syndrome
Pfeiffer first described this syndrome in 1964 as acrocephalosyndactyly syndrome, comprising bicoronal craniosynostosis, midfacial hypoplasia, broad thumbs and great toes, and partial and variable soft tissue syndactyly of the hands and feet.1
The syndrome is classified into 3 subtypes:
• Type 1: a mild presentation, with brachycephaly, midfacial hypoplasia, and finger and toe deformities; generally associated with a good outcome with normal intelligence.
• Type 2: cloverleaf-shaped skull, severe proptosis, finger and toe deformities, elbow ankylosis, neurologic complications with delay in development, and various visceral anomalies; has a poor prognosis, with death by 2 years.2-4
• Type 3: same as type 2, but with no cloverleaf skull deformity.

Pathogenesis
Pfeiffer syndrome results from mutations in the FGFR1 or FGFR2 genes, which encode fibroblast growth factor receptors types 1 and 2, respectively. FGFR1 plays an important role in cell signaling, and a mutation can enhance early maturation of bone cells and premature fusing of the skull, hands, and feet. FGFR2 encodes for a protein that has a role in cell division and regulation of cell growth and maturation.3 Mutations in this gene affect embryonic development and are more frequently the cause of Pfeiffer syndrome.3 More than 40 different mutations in FGFR2 have been identified as causing Pfeiffer syndrome.3 Autosomal dominance with complete penetrance is characteristic, despite variable expressivity.1
It has been speculated that sporadic cases of Pfeiffer syndrome have been related to advanced paternal age,4 which might be the case in our patient.
This syndrome affects approximately 1 in 120,000 births.3 Along with the classic picture of craniosynostosis, syndactyly, and broad, radially deviated thumbs, Pfeiffer syndrome occasionally features hydrocephaly, ocular proptosis, elbow ankylosis, and abnormal viscera.2 Slow development and early death is not unusual for children with types 2 and 3 of the condition.
Diagnosis
Craniosynostosis can be associated with various syndromes (Table). The distinguishing feature of Pfeiffer syndrome compared with other causes of early skull fusion is the presence of broad thumbs and great toes.
The diagnosis of Pfeiffer syndrome type 2 can be made based on distinctive features such as the cloverleaf head and the broad thumbs and toes. In our case, genetics was consulted and made the final diagnosis. Molecular genetic testing also could be performed to confirm the clinical diagnosis.
Management
It is recommended that the synostotic sutures be surgically released during the first year, often as early as at 3 months of age, to decompress the brain. A procedure to elongate and expand the bony orbits also can be done during the first year. As the child grows, further interventions and skull remodeling might be required to reduce the risk of hydrocephaly.
A second stage of management is midfacial surgery to reduce the exophthalmos and to correct midfacial hypoplasia.
It also is necessary to add artificial tear ointment to prevent damage from dry eyes resulting from proptosis. Maxillary hypoplasia could lead to a small larynx and pharynx, which in turn can lead to respiratory distress and the possible aspiration of food into the lungs. In our patient, a gastrostomy tube was placed to prevent this from occurring. n
Hannah Lee, DO, is a first-year resident in the Department of Pediatrics at the University of Texas at Houston Health Science Center at Houston.
Emma A. Omoruyi, MD, MPH, is an assistant professor of pediatrics at the University of Texas at Houston Health Science Center.
References
1. Oyamada MK, Ferreira HS, Hoff M. Pfeiffer syndrome type 2 – case report. Sao Paulo Med J. 2003;121(4):176-179.
2. Vogels A, Fryns J-P. Pfeiffer syndrome. Orphanet J Rare Dis. 2006;1:19.
3. Jay S, Wiberg A, Swan M, et al. The fibroblast growth factor receptor 2 p.Ala172Phe mutation in Pfeiffer syndrome—history repeating itself. Am J Med Genet A. 2013;161A(5):1158-1163.
4. Park MS, Yoo JE, Chung J, Yoon SH. A case of Pfeiffer syndrome. J Korean Med Sci. 2006;21(2):374-378.