
Headache and Neck Pain in Young Boy Who Drags Foot While Walking
Photo Finish
Acute Dx: What Cause of Sudden Illness?
THE CASE: An 8-year-old boy presents to your office with persistent frontal headaches and diffuse neck pain of 2 weeks’ duration. His mother reports that for the past week he had been dragging his right foot while walking. The headache intensity is unaffected when treated with ibuprofen or acetaminophen. The child attends public school. He has been healthy but less active and more sleepy than usual.
Vital signs are stable. The physical examination reveals weakness of the right side of his neck and decreased strength in all muscle groups of the right upper and lower extremities. Reflexes and sensation are intact. Asymmetry of gait is noted with ambulation. Remaining neurological findings are nonfocal.
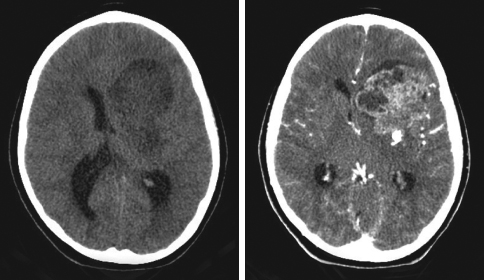
The child is referred to the local emergency department for further evaluation. CT scans of the head with and without contrast are shown.
What do you suspect is the most likely diagnosis?
• Anaplastic astrocytoma
• Ependymoma
• Metastasis to the CNS
• Myxoid chondrosarcoma
• Hemorrhagic rhabdomyosarcoma
(Answer and discussion on next page)
Photo Finish
Answer: Myxoid chondrosarcoma
DISCUSSION: The CT scans showed a large (7.5 3 5-cm) heterogeneously enhancing mass in the left frontotemporal lobe with focal areas of necrosis. A significant mass effect with effacement of the left lateral ventricle—a 10-mm midline shift—was also present. The patient was referred to a neurosurgery team and admitted to the hospital. Treatment with intravenous dexamethasone was started, and MRI was performed (Figure). The next day, he underwent craniotomy and resection of the tumor. The resected pink-tan mass was sent for immunohistochemical analysis. The final surgical pathology results were consistent with primary intracranial myxoid chondrosarcoma.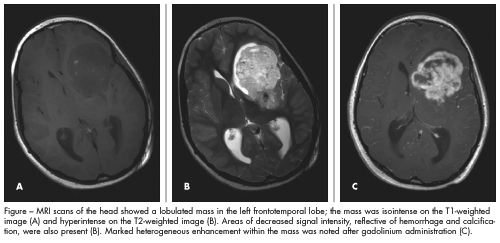
Primary intracranial extraosseous chondrosarcoma is a rare neoplasm. It is often attached to the dura; typical locations include the cerebral convexity, falx, fourth ventricle, cerebellum, and thalamus. The tumor is believed to arise from the mesenchymal components of the CNS—the primitive multipotential mesenchymal cells or their mature descendents (fibroblasts, meningeal cells, and pial cells). In contrast, primary cranial chondrosarcoma normally arises from the skull base, and intraparenchymal chondrosarcoma is thought to originate from the misplaced embryonal cartilage rests or primitive multipotential mesenchymal cells in leptomeningeal sheaths around vessels or vessels walls.
No definitive treatment recommendations exist for intracranial chondrosarcoma. Radical excision is thought to play an important role in the management of this tumor. Postoperative radiotherapy to prevent recurrence and progression also must be considered.
In this child, expressive aphasia developed postoperatively, and he was discharged to a rehabilitation facility. At follow-up, he was noted to have improvements in both motor strength on his right side and speech. He was scheduled to receive a total of 6 courses of chemotherapy with ifosfamide and etoposide.
DIFFERENTIAL DIAGNOSIS
Primary CNS tumors are the second most frequent malignancy in childhood and adolescence. The overall mortality rate among this group is 45%. The tumors are generally classified according to the World Health Organization (WHO) classification system, which takes into account the cell of origin of the tumor and the tumor location. The following tumors could be responsible for this child’s presentation.
Gliomas are a group of CNS tumors that arise from glial cells—the cells that support the neurons that make up the normal brain. WHO grade I gliomas tend to be the least aggressive and WHO grade IV gliomas the most aggressive and malignant. High-grade gliomas encompass WHO grade III gliomas (anaplastic astrocytoma) and WHO grade IV gliomas (glioblastoma multiforme).
Astrocytomas comprise 40% of pediatric CNS tumors. Astrocytomas in childhood are predominantly low-grade. Juvenile pilocytic astrocytoma, the most common astrocytoma in children, can occur anywhere in the CNS; however, the classic site is within the cerebellum. This tumor has low metastatic potential and is rarely invasive.
Malignant astrocytomas are much less common in children and adolescents than in adults. Among this group, anaplastic astrocytoma is much more common than glioblastoma multiforme. Clinical management of astrocytoma is multimodal in that it incorporates surgery as the primary treatment as well as radiation and chemotherapy.
Oligodendrogliomas are uncommon tumors of childhood. They occur predominantly in the cerebral cortex and originate in the white matter. In a patient who presents with seizure, observation of a calcified cortical mass on a CT scan is suggestive of oligodendroglioma. Treatment approaches for oligodendrogliomas are similar to those for infiltrating astrocytomas.
Ependymomas account for 10% of childhood tumors. These usually noninvasive tumors may extend into the ventricular lumen or displace the normal structures. The most common site of origin is the posterior fossa. Surgery is the primary treatment modality. The extent of surgical resection, patient age, and tumor location are major prognostic factors. Poorer outcomes are associated with younger age and tumors located in the posterior fossa. Radiation therapy of the involved field is recommended for most children with ependymomas.
Embryonal tumors. Primitive neuroectodermal tumors (PNETs) are the most common group of malignant CNS tumors of childhood. Although the most frequent site of origin of PNETs is the cerebellar vermis, about 5% of these tumors can be supratentorial. Medulloblastoma accounts for 90% of embryonal tumors; it occurs in the cerebellum, predominantly in boys at a median age of 5 to 7 years. PNET or medulloblastoma often manifests as obstructive hydrocephalus and increased intracranial pressure. Signs and symptoms include headache, nausea, vomiting, drowsiness and other behavior changes, and ataxia. These clinical characteristics are often indistinguishable from those of other posterior fossa tumors, including ependymoma and cerebellar astrocytoma.
The 3 aims in the management of PNETs are: assessment and treatment of increased intracranial
pressure, complete or near-complete resection of the tumor, and reestablishment of the normal cerebrospinal fluid flow.
Metastasis. Metastasis of other childhood malignancies to the brain is rare. Childhood acute lymphocytic leukemia and non-Hodgkin lymphoma can spread to the leptomeninges and cause symptoms of communicating hydrocephalus. Chloromas can develop throughout the neuraxis. Brain parenchymal metastasis occurs from neuroblastoma, rhabdomyosarcoma, Ewing sarcoma, osteosarcoma, clear cell sarcoma of the kidney, and chondrosarcoma. Therapy depends on the specific histological diagnosis and may include radiation therapy, intrathecal chemotherapy, and systemic chemotherapy. ■
FOR MORE INFORMATION:
■ Blaney SM, Kun LE, Hunter J, et al, eds. Tumors of the central nervous system. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:786-864.
■ IM SH, Kim DG, Park IA, Chi JG. Primary intracranial myxoid chondrosarcoma: report of a case and review of the literature. J Korean Med Sci. 2003;18:301-307.
■ Korten AG, ter Berg HJ, Spincemaille GH, et al. Intracranial chondrosarcoma: review of the literature and report of 15 cases. J Neurol Neurosurg Psychiatry. 1998;65:88-92.
■ Kuttesch JF Jr, Ater JL. Brain tumors in childhood. In: Kliegman RM, Behrman RE, Jenson HB, Stanton BM, eds. Nelson Textbook of Pediatrics. 18th ed. Philadelphia: Elsevier; 2007:chapter 497.
■ Maity A, Pruitt AA, Judy KD, et al. Cancer of the central nervous system. In: Abeloff MD, Armitage JO, Niederhuber JE, et al, eds. Abeloff’s Clinical Oncology. 4th ed. Philadelphia: Elsevier; 2008:1075-1136.