
A Girl With Short Stature: Which Gene(s) Is It?
Genetic Disorders
An 11-year-old girl presented with short stature and a deformity of her left upper arm (Figure 1). Her past medical history included 2 fractures of the left humerus. Her family history was positive for short stature in the mother and father, whose heights were 147 cm and 150 cm, respectively.

Physical examination results were significant for height below the third percentile (50th percentile for 8½ years of age) and weight below the third percentile (50th percentile for 8 years of age). She had triangular facies, blue sclerae, grayish discoloration of her teeth, bowing of the left humerus, and a café au lait macule on the left leg (Figure 2). She was prepubertal.

Laboratory testing was performed for karyotype, growth hormone deficiency, hypothyroidism, osteogenesis imperfecta (OI), and a short stature homeobox (SHOX) gene mutation.
What diagnosis (or diagnoses) would you suspect?
ANSWER: Osteogenesis imperfecta type I and polyostotic fibrous dysplasia
Laboratory test results showed a normal karyotype, an insulinlike growth factor level of 110 ng/mL (reference range, 117-771 ng/mL), and a free thyroxine level of 0.6 ng/dL (reference range, 0.6-1.6 ng/dL). A mutation was found in the collagen type I α2 gene (COL1A2) on chromosome 7, confirming a diagnosis of OI type I. Test results for SHOX mutation were negative.
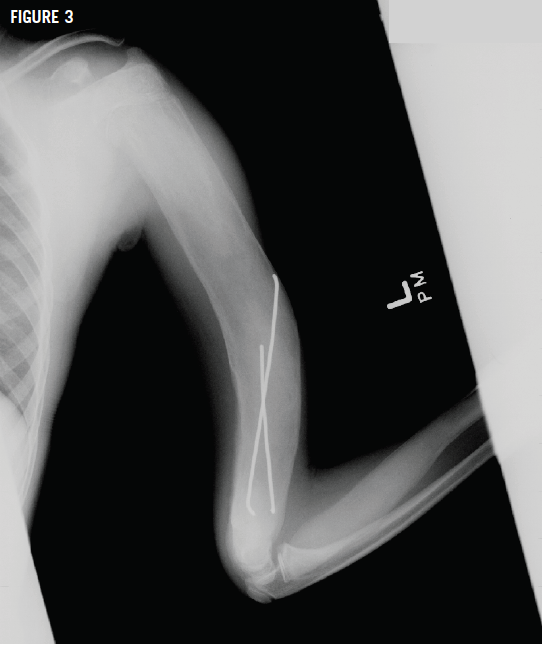
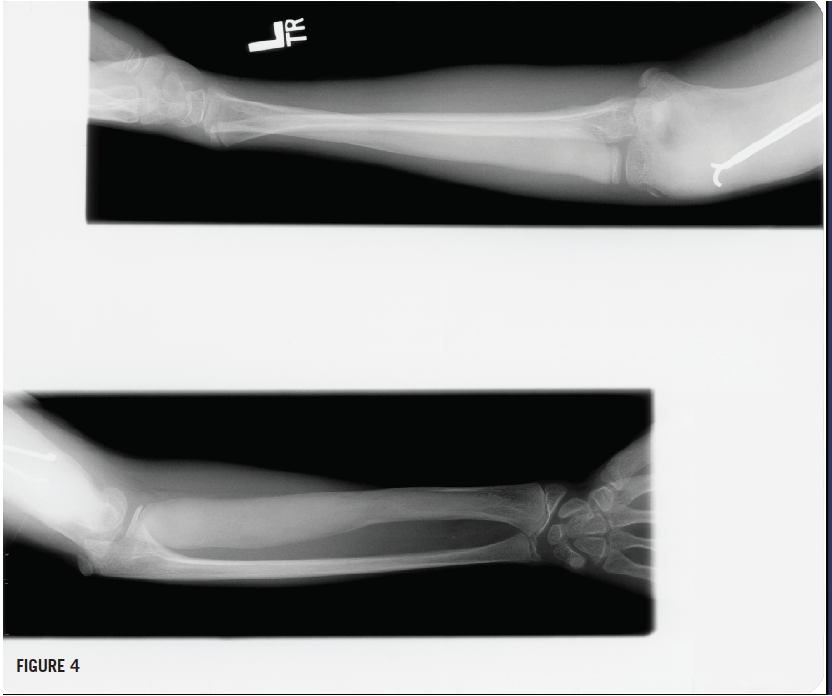
Radiographs of the left humerus showed lesions with ground-glass opacity and a Madelung deformity of the wrist (Figures 3 and 4), suggesting polyostotic fibrous dysplasia (FD). Bone age and chronologic age were similar. Results of magnetic resonance imaging of the brain were normal.
Thyroid replacement and recombinant growth hormone (rGH) injections were started.
DIFFERENTIAL DIAGNOSIS
In addition to familial FD and OI, the differential diagnosis of our patient’s short stature included growth hormone deficiency, hypothyroidism, and a SHOX gene mutation with associated Madelung deformity. However, because of the patient’s FD in an upper extremity, only slightly abnormal growth hormone levels and thyroid function, and negative SHOX test results, the latter 3 etiologies were less likely.
OSTEOGENESIS IMPERFECTA
OI is a hereditary disorder caused by a defect in collagen synthesis. It affects 1 in 20,000 persons. The responsible genes are located on chromosomes 7 (COL1A2) and 17 (COL1A1).1 A diagnosis of OI explains our patient’s short stature, triangular facies, blue sclerae, and dentinogenesis imperfecta. Scoliosis secondary to collapsed vertebrae, curved lower-extremity bones secondary to muscle imbalances, and fractures through epiphyseal growth plates also can negatively affect growth in children with OI. Bisphosphonates are the mainstay of treatment, but rGH therapy is promising.2,3
FIBROUS DYSPLASIA
FD is a nonhereditary disorder in which normal bone is replaced by fibrous tissue. It affects 1 in 10,000 persons. The responsible gene, which codes for the G-protein α subunit (GNAS), is located on chromosome 20 and is responsible for the production of cyclic adenosine monophosphate.4 The resultant stimulation of melanocytes and osteoblasts explains our patient’s café au lait macule and arm deformity.
This same mutation occurs in McCune-Albright syndrome (MAS), with studies suggesting that small mutations in the gene result in MAS and large mutations in FD.5 A diagnosis of MAS is generally considered to require 2 of the following 3 features: polyostotic FD, extensive café au lait macules with irregular borders, and hypersecretory endocrinopathies.6
Our patient, however, had a single, smooth-edged macule rather than multiple, irregularly edged macules, and she had endocrine hypofunction abnormalities, not hypersecretion.
Case Follow-up
Now at age 17, our patient continues on daily thyroid replacement and a weekly oral bisphosphonate. The rGH was discontinued at age 15, when she was skeletally mature. Now at 152 cm, she has surpassed the height of both of her parents.
REFERENCES:
1. Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7(9):540-557.
2. Marini JC, Bordenick S, Heavner G, Rose S, Chrousos GP. Evaluation of growth hormone axis and responsiveness to growth stimulation of short children with osteogenesis imperfecta. Am J Med Genet. 1993;45(2):261-264.
3. Antoniazzi F, Monti E, Venturi G, et al. GH in combination with bisphosphonate treatment in osteogenesis imperfecta. Eur J Endocrinol. 2010;163(3):479-487.
4. Singer FR. Fibrous dysplasia of bone: the bone lesion unmasked. Am J Pathol. 1997;151(6):
1511-1515.
5. Cohen MM Jr, Howell RE. Etiology of fibrous dysplasia and McCune-Albright syndrome. Int J Oral Maxillofac Surg. 1999;28(5):366-371.
6. Madsen H, Borges MT, Kerr JM, Lillehei KO, Kleinschmidt-Demasters BK. McCune–Albright syndrome: surgical and therapeutic challenges in GH-secreting pituitary adenomas. J Neurooncol. 2011;104(1):215-224.