
Peer Reviewed
Autosomal Dominant Polycystic Kidney Disease
Authors:
Gaby S. Gil, MD; Mohammad Saeed, MD; and Shobhana Chaudhari, MD
New York Medical College at Metropolitan Hospital Center, New York, New York
Citation:
Gil GS, Saeed M, Chaudhari S. Autosomal dominant polycystic kidney disease. Consultant. 2017;57(10):616-617.
An 87-year-old man presented with a gradual onset of increased abdominal girth of 1 year’s duration. He reported that the associated symptoms had progressed to constant abdominal pain, fatigue, and decreased exercise tolerance. Over time, he noticed that he had become slower than usual in performing his daily activities.
History and physical examination. The patient’s medical history was remarkable for stage 3 chronic kidney disease and hypertension; he also had a history of smoking. The initial physical examination findings showed a blood pressure of 140/60 mm Hg, a pulse rate of 68 beats/min and regular, a respiratory rate of 20 breaths/min, and a temperature of 37.2°C. There was no jugular vein distention, no carotid bruits, and no cardiac murmurs. His lungs were clear. Abdominal examination findings were remarkable for palpable nontender masses over the flanks and hepatomegaly of 4 cm below the right costal margin. No ascites, no fluid wave, and no collateral circulation were present.
Diagnostic tests. Results of a basic metabolic panel, including levels of potassium, sodium, magnesium, calcium, phosphorus, and albumin, were within normal limits. Renal function test results included a blood urea nitrogen level of 27 mg/dL, a serum creatinine level of 1.4 mg/dL, and an estimated glomerular filtration rate (GFR) of 48 mL/min.
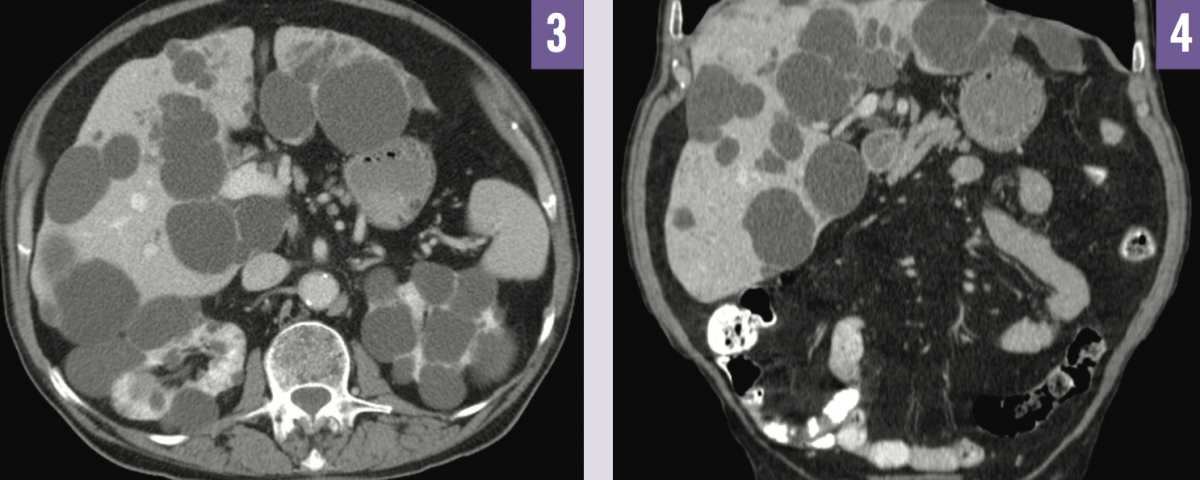
Abdominal ultrasonography demonstrated bilateral enlarged kidneys with several cysts of varying size (Figure 1), and multiple scattered echo-dense cysts measuring up to 7 cm throughout the liver (Figure 2). Noncontrast computed tomography (CT) imaging showed numerous hepatic cysts in association with bilateral kidney cysts (Figures 3 and 4). These findings were compatible with a radiographic diagnosis of autosomal dominant polycystic kidney disease (ADPKD).

NEXT: Outcome of the Case
Outcome of the case. The patient’s clinical course was uneventful. His hypertension is controlled on a daily oral regimen of hydrochlorothiazide, 25 mg; amlodipine, 10 mg; and carvedilol, 25 mg. His initial medication regimen had included oral lisinopril, 5 mg/d, but he developed elevated serum potassium levels up to 6.4 mEq/L, and this approach was discontinued. The patient has not experienced kidney or liver cyst complications. His serum creatinine level and GFR have been stable at 1.3 mg/dL and 52 mL/min, respectively.
Discussion. ADPKD is a commonly encountered late-onset multisystem illness with a variety of manifestations and severity of symptoms. Approximately 85% of individuals with ADPKD have mutation of PKD1 on chromosome 16 (ADPKD type 1), and the remaining 15% of cases involve mutation of PKD2 on chromosome 4 (ADPKD type 2).1 These genes encode proteins that have a role in the regulation and proliferation of renal tubular epithelial cells.1 ADPKD typically affects persons in the third or fourth decade of life and less commonly affects persons aged 65 years or older.2 More than 50% of persons with ADPKD type 1 develop end-stage kidney disease requiring dialysis by age 50, compared with a mean age of 70 years for persons with ADPKD type 2, which is the milder form of the disorder.2
ADPKD may produce an array of clinical manifestations such as dull aching flank pain due to increased cystic size, kidney stones, or complications related to cyst infection, hemorrhage, or rupture. Other kidney manifestations can include renal insufficiency, hematuria, urinary tract infection, and decreased urine-concentrating ability. All these complications are directly related to the extent of renal cyst size. Extrarenal presentation of the disease is possible and can involve cardiac murmur due to mitral valve prolapse; abdominal wall hernias; pancreatic, hepatic, or splenic cysts; diverticulosis; and brain aneurysm.1,2
On physical examination, the most common features include elevated blood pressure early in the course of ADPKD, even when renal function is still normal, and palpable bilateral flank masses or nodular hepatomegaly.1,3
The diagnosis of ADPKD is made through renal ultrasonography due to its readiness, cost-effectiveness, and low level of radiation exposure. The following ultrasonographic criteria are used for diagnosis, based on age and the number or renal cysts: the presence of at least 3 unilateral or bilateral kidney cysts in individuals aged 15 to 39 years; at least 2 cysts in each kidney in individuals aged 40 to 59 years; and at least 4 cysts in each kidney in individuals aged 60 years or older.4
In certain circumstances, contrast-enhanced abdominal CT scan or magnetic resonance imaging may help identify smaller cysts in individuals with no contraindications for contrast media.4
The primary cause of mortality in ADPKD is of cardiac origin. Other causes of mortality include infection and ruptured intracranial aneurysm, the latter of which represents the most serious complication.1 The estimated prevalence of cerebral aneurysms is near 5% among the younger population, but the prevalence increases with age, reaching as high as 20% in individuals aged 60 years or older.1-3 The mortality rate is approximately 10% in patients below age 50 years with poorly controlled hypertension.1-3 Routine screening with magnetic resonance angiogram without gadolinium is tailored for high-risk patients, such as those with a personal history of ruptured cerebral aneurysm, a strong family history of hemorrhagic cerebrovascular accident secondary to subarachnoid hemorrhage, warning symptoms such as acute onset of severe headache, and a high-risk occupation.5-8
The mainstay of treatment for all patients with ADPKD consists of blood pressure control with renin-angiotensin-aldosterone system blockers (eg, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, direct renin inhibitors). These are the preferred agents due to their well-established role in the prevention of cardiovascular disease. Vasopressin V2-receptor antagonists (eg, tolvaptan) have demonstrated beneficial effects in reducing the cyst burden and protecting kidney function in ADPKD, as reported in the TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes) trials.9,10
Supportive measures such as decreased dietary sodium intake, maintenance of hydration, and management of chronic abdominal pain, as well as treatment of nephrolithiasis and cyst infection with trimethoprim-sulfamethoxazole or fluoroquinolones, are also an essential part of management.5-7 Ultimately, individuals who progress to end-stage kidney disease require renal replacement therapy or renal transplant.11
REFERENCES:
- Akoh JA. Current management of autosomal dominant polycystic kidney disease. World J Nephrol. 2015;4(4):468-479.
- Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329(5):332-342.
- Grantham JJ. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008;359(14):1477-1485.
- Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205-212.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009;20(9):1888-1893.
- Chapman AB, Bost JE, Torres VE, et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2012;7(3):479-486.
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149-168.
- Schievink WI, Torres VE, Piepgras DG, Wiebers DO. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1992;3(1):88-95.
- Torres VE, Chapman AB, Devuyst O, et al; TEMPO 3:4 Trial Investigators. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407-2418.
- Higashihara E, Torres VE, Chapman AB, et al; TEMPO42 and 156-05-002 Study Investigators. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol. 2011;6(10):2499-2507.
- Hadimeri H, Johansson AC, Haraldsson B, Nyberg G. CAPD in patients with autosomal dominant polycystic kidney disease. Perit Dial Int. 1998;18(4):429-432.
1. Wilson PD. Polycystic kidney disease. N Engl JMed. 2004;350:151-164.