
A Collection of Conditions Related to Autoimmunity
Granulomatosis With Polyangiitis
Laura E. Bateman, MD
Ochsner Medical Center, New Orleans, Louisiana
A 66-year-old woman presented to the emergency department (ED) with a 2-day history of dark urine and shortness of breath. She also reported a number of recent visits to the ED and clinic for new-onset asthma and leg pain.
History. Over the past year, the patient had been started on an albuterol inhaler for asthma. Approximately 4 months before the current presentation, she had developed acute-onset, bilateral sensorineural hearing loss. Results of a magnetic resonance imaging scan of the brain at that time were unremarkable. Within 2 months, she had experienced progressive ankle swelling, with pain in the bilateral lower extremities. One month later, she presented to the ED with shortness of breath, which was treated as an asthma exacerbation. While in the ED, she developed right-sided facial droop consistent with Bell palsy. A stroke workup did not elucidate any further etiology. Corticosteroids were administered, which improved both the facial palsy and the leg pain.
Physical examination. Physical examination revealed a well- developed woman with mild alopecia. On arrival, her temperature was 37.8°C, heart rate was 93 beats/min, and blood pressure was 182/95 mm Hg.
Pertinent findings included mild wheezing in the right lung fields on expiration without crackles or rales.
Neurologic examination showed light-touch sensory loss in a stocking-glove distribution and mild right facial droop, particularly at the right nasolabial fold, with forehead involvement. Results of a Weber test were positive for hearing loss. Her lower extremities were tender to palpation with a shiny appearance and 1+ pitting edema to just above the ankles bilaterally.
Diagnostic tests. The hemoglobin concentration was decreased at 8.3 g/dL, with a mean corpuscular volume of 85 µm3 and a platelet count of 664 × 103/µL. The erythrocyte sedimentation rate was elevated at 85 mm/h. Blood urea nitrogen level was 39 mg/dL, and creatinine level was 3.2 mg/dL. Results of a comprehensive metabolic panel were pertinent for an albumin level of 2.1 g/dL and a bicarbonate level of 18 mEq/L. Electrolyte levels and liver function test values were normal.
Urinalysis was positive for large amounts of protein and blood. Visualization of the urine sediment showed muddy brown casts and dysmorphic red blood cells.
Chest radiographs revealed 3 nodules in the patient’s right middle lung.

Diagnosis. The patient received a diagnosis of granulomatosis with polyangiitis (GPA), also called Wegner granulomatosis. During her hospitalization, test results returned positive for proteinase 3 cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA/PR3). Kidney biopsy findings under light microscopy (Figures 1A and 1B) demonstrated cellular crescents with fibrinoid necrosis, along with mild to moderate interstitial lymphocytic infiltrates with overall 5% interstitial fibrosis, findings consistent with pauci-immune necrotizing crescentic glomerulonephritis, confirming the diagnosis of GPA.
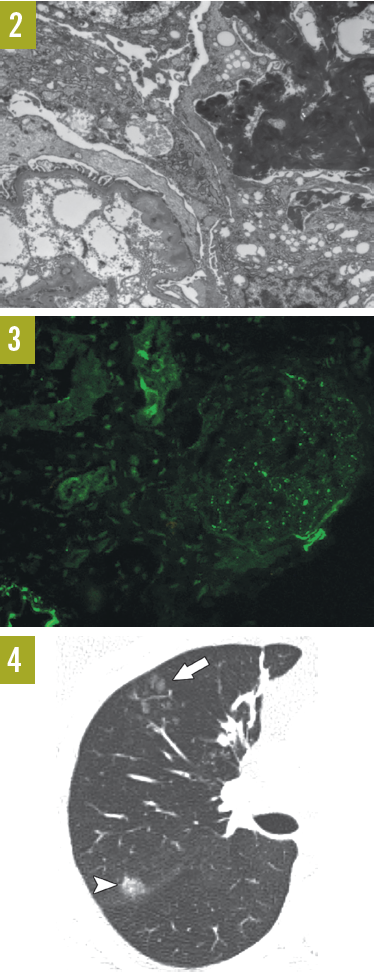
Toluidine blue staining of the kidney biopsy under electron microscopy (Figure 2) showed no subepithelial or subendothelial immune deposits. Visceral epithelial cell foot-process effacement was present. Endothelial cells were noted to be filled with fibrinoid material. Immunofluorescence microscopy of the kidney biopsy (Figure 3) demonstrated positive arteriolar and mesangial staining for C3. High-resolution computed tomography scanning of the right lung (Figure 4) showed centrilobular ground-glass attenuation nodules (arrow) and larger subpleural ground-glass nodule (arrowhead).
Discussion. GPA is a vasculitis of small vessels characterized by the presence of necrotizing granulomas. The classic triad of organ involvement is that of sinus, respiratory tract, and kidneys. However, as with other systemic autoimmune diseases, the organ systems involved can be extremely variable, leading to a delay in diagnosis due to the apparently incongruous nature of the symptoms. Biopsy results combined with a compatible clinical picture and seropositivity for c-ANCA are diagnostic.
On the list of differential diagnoses are Churg-Strauss syndrome (eosinophilic granulomatosis), Goodpasture syndrome, and sarcoidosis.
Although Churg-Strauss syndrome can present with pulmonary infiltrates, it typically does not develop in to an acute glomerulonephritis, as in our patient’s case. Symptoms of the disease may include asthma, atopy, neuropathies, and hypereosinophilia, which in turn causes tissue damage, most commonly to the lungs and the digestive tract. Serum abnormalities may include eosinophil levels greater than 10% of a differential white blood cell count, and perinuclear ANCA positivity.
While Goodpasture syndrome is similar to GPA, neurologic symptoms are rare. In Goodpasture syndrome, antibodies attack the ɑ-3 subunit of type IV collagen in the basement membrane in lungs and kidneys, leading to bleeding from the lungs and to kidney failure.
Sarcoidosis also can be a multisystem vasculitic disease; however, it is not commonly associated with glomerulonephritis, and biopsy results typically will show well-differentiated noncaseating granulomas.
Untreated, GPA is uniformly fatal. Renal and pulmonary involvement can progress rapidly, and therefore this syndrome warrants urgent treatment. While the ANCA-associated vasculitides can be life-threatening, treatment can be highly effective. Between 70% and 90% of patients with GPA attain clinical remission with initial immunosuppressive therapy. Treatment regimens may include corticosteroids or immunosuppression with agents such as cyclophosphamide or rituximab. Long-term management on corticosteroid-sparing agents such as methotrexate must address not only the induction of remission but also the potential need for repeated courses of therapy, since relapse is common.
Outcome of the case. Pulse-dose corticosteroids and plasmapheresis were initiated, followed by a course of cyclophosphamide. The patient remained in remission at 6 months on methotrexate.
NEXT: Cutaneous Granulomatosis With Polyangiitis
Cutaneous Granulomatosis With Polyangiitis
Jennifer F. Gregory, MD
Naval Flight Surgeon, Carrier Air Wing THREE,
Naval Air Station Oceana, Virginia Beach, Virginia
Trisha C. Beute, MD
Atlantic Dermatology Associates, Virginia Beach, Virginia
A 59-year-old man presented with red, tender papules on his back that had developed after he had begun a prednisone taper. He had taken prednisone daily for some time for multiple systemic symptoms.
History. The patient had been in the care of a rheumatologist, a nephrologist, and a pulmonologist for granulomatosis with polyangiitis (GPA). His symptoms at the time of diagnosis had included persistent nasal crusting and discharge, dyspnea, hemoptysis, arthralgia, oliguria, and dysuria. Laboratory test results at the time of diagnosis had included a cytoplasmic antineutrophil cytoplasmic antibody (c-ANCA) screening titer greater than 1:640, an erythrocyte sedimentation rate (ESR) of 79 mm/h, and a C-reactive protein (CRP) level of 69 mg/L. Bronchoscopy and renal biopsy results did not reveal characteristic histologic findings.
Physical examination. Physical examination revealed approximately 10 erythematous papules, measuring 2 to 3 mm each, scattered across the patient’s back, which were tender to palpation (Figure 1).

Figure 1. Scattered tender erythematous papules on the patient’s back.

Figure 2. Left, acneiform lesion with follicular dilatation and parakeratotic plugging but with no vasculitis (H&E; Å~10 magnification). Right, perifollicular granulomatous inflammation and multinucleated histiocytes (H&E, Å~20 magnification).
Diagnostic tests. A punch biopsy of a single papule was performed; hematoxylin and eosin (H&E) stained sections are shown in Figure 2. Histopathologic examination revealed an acneiform lesion with follicular dilatation and parakeratotic plugging. Perifollicular granulomatous inflammation and multinucleated histiocytes were seen. There was no vasculitis.
Based on the history, physical examination findings, and laboratory test results, the man received a diagnosis of GPA.
Treatment. Intralesional triamcinolone acetonide, 10 mg/mL, was injected into 4 lesions, 0.1 mL/lesion. All of the patient’s lesions have since resolved.
Discussion. GPA is an ANCA-associated vasculitis affecting small to medium-sized vessels. It is characterized by necrotizing granulomas of the upper and lower respiratory tracts and glomerulonephritis.
Most patients are between 40 and 50 years old at onset, with a male predominance, although pediatric cases have been reported.1 Patients most commonly present with upper respiratory tract symptoms such as rhinorrhea, chronic sinusitis, and nasal discharge.
Approximately 45% of patients have cutaneous findings secondary to vasculitis or extravascular granulomatous inflammation, with greater prevalence among patients with generalized GPA compared with limited disease.2,3 The most common cutaneous manifestation is palpable purpura; other findings include subcutaneous nodules, ulcers, or pustular eruptions. Lesions previously diagnosed as “malignant pyoderma” now are recognized as necrotizing angiitis of GPA.4 Rare cases of acneiform lesions due to extravascular granulomatous inflammation have been reported, particularly in the absence of vasculitis.1,4
Histopathologically, cutaneous GPA may manifest as leukocytoclastic vasculitis (LCV), extravascular granuloma, or a mixed inflammatory pattern.3 LCV often correlates with palpable purpura, extravascular granulomas with nodular lesions, and mixed inflammatory pattern with necrotizing ulcerations. The differential diagnosis of vascular purpuric lesions includes other ANCA-associated vasculitides (microscopic polyangiitis, Churg-Strauss syndrome), immune complex-mediated vasculitis, collagen tissue disease (eg, Sjögren syndrome), rickettsial and viral infections, and malignancy-associated vasculitis.5 The differential diagnosis of granulomatous lesions includes sarcoidosis and rosacea. The unique histologic feature of our patient’s biopsy was the presence of granulomatous inflammation in the absence of vasculitis.
Among patients with active systemic GPA, 75% to 80% have elevated c-ANCA (antiproteinase-3) titers during their disease course.2,4 Specific antibody detection allows for earlier diagnosis, treatment, and improved prognosis. Untreated patients have a 5-month mean survival and a 90% 2-year mortality, most often due to renal failure.
Treatment of GPA traditionally consisted of immunosuppression with high-dose prednisone and cyclophosphamide, methotrexate, or azathioprine.2 Newer corticosteroid-sparing immunomodulators, such as rituximab, are particularly effective in severe or refractory cases.6-8 Nearly 93% of patients who receive treatment achieve remission and average 4 years prior to relapse.2 Staphylococcus aureus carriage has been associated with an increased relapse risk, and patients may benefit from long-term trimethoprim-sulfamethoxazole therapy.9,10
Outcome of the case. At follow-up, the patient was in remission after 2 courses of rituximab, with a negative c-ANCA titer, an ESR of 10 mm/h, and CRP level below 5 mg/L. His respiratory and renal symptoms had significantly improved, and his maintenance therapy consisted of daily azathioprine with a very gradual prednisone taper.
References:
- Brazzelli V, Vassallo C, Baldini F, Ravelli A, Martini A, Borroni G. Wegener granulomatosis in a child: cutaneous findings as the presenting signs. Pediatr Dermatol. 1999;16(4):277-280.
- James WD, Berger TG, Elston DM. Cutaneous vascular diseases. In: James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Saunders Elsevier; 2011:chap 35.
- Daoud MS, Gibson LE, DeRemee RA, Specks U, el-Azhary RA, Su WP. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31(4):605-612.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener’s granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34(10):739-747.
- Chen KR, Carlson JA. Clinical approach to cutaneous vasculitis. Am J Clin Dermatol. 2008;9(2):71-92.
- Cartin-Ceba R, Golbin JM, Keogh KA, et al. Rituximab for remission induction and maintenance in refractory granulomatosis with polyangiitis (Wegener’s): ten-year experience at a single center. Arthritis Rheum. 2012;64(11):3770-3778.
- Specks U, Merkel PA, Seo P, et al; RAVE-ITN Research Group. Efficacy of remission-induction regimens for ANCA-associated vasculitis. N Engl J Med. 2013;369(5):417-427.
- Stone JH, Merkel PA, Spiera R, et al; RAVE-ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221-232.
- Stegeman CA, Tervaert JWC, de Jong PE, Kallenberg CGM; Dutch Co-trimoxazole Wegener Study Group. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. N Engl J Med. 1996;335(1):16-20.
- Tadema H, Heeringa P, Kallenberg CGM. Bacterial infections in Wegener’s granulomatosis: mechanisms potentially involved in autoimmune pathogenesis. Curr Opin Rheumatol. 2011;23(4):366-371.
Acknowledgement and Disclaimer:
The authors thank Tim Sorrells, MD, and Bruno Schmitz, MD, in the Pathology Department at Naval Medical Center Portsmouth, Virginia, for the histology photographs.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, the Department of Defense, or the US Government.
NEXT: Systemic Lupus Erythematosus Presenting With Raynaud Phenomenon
Systemic Lupus Erythematosus Presenting With Raynaud Phenomenon
S. Rafik Bouaziz, MD; Ishwinder Singh Sehgal, MD; Himika Dalia, MD; and Gregory Karamian
St. George’s University, Grenada

A 48-year-old woman with 15 pack-year history of cigarette smoking presented with swollen, erythematous, and extremely painful fingers (Figures 1-4). She had been taking oral levofloxacin for community-acquired pneumonia, diagnosed 4 days prior. She had taken levofloxacin before without adverse reaction. Mild pain in her fingertips had begun prior to her first dose of levofloxacin.
History. She denied a personal or family history of autoimmune disease. She had had one episode of Raynaud phenomenon (RP) in the remote past, but during that episode, her fingers had been cold, pale white, and numb.
The woman had chronic, diffuse pain syndrome and took combination buprenorphine (8 mg) and naloxone (2 mg) sublingually 3 times daily. Her other medications were escitalopram, 10 mg once daily, and tiagabine, 8 mg before bed. She also used a combination estradiol/norethindrone acetate (0.62/2.7 mg) transdermal patch.

Diagnostic tests. Laboratory studies disclosed the following values: extractable nuclear antigen, positive; C-reactive protein, 63.5 mg/L (reference range, 0.0-8.0 mg/L); antinuclear antibody (ANA), positive; antiphospholipid immunoglobulin M (IgM) antibodies, 15.9 MPL (reference range, 0.0-14.9 MPL); and antiphospholipid immunoglobulin G (IgG) antibodies, 65.3 GPL (reference range, 0.0-14.9 GPL).
Treatment. Her use of the estradiol/norethindrone acetate transdermal patch was immediately stopped. She was started on sustained-release nifedipine, 30 mg once daily, and topical nitroglycerin in small amounts applied to her fingers twice daily. Levofloxacin was continued, and she was scheduled for a follow-up appointment in 10 days.
Upon follow-up, her pneumonia had not improved significantly, so she was directly admitted to the hospital from the outpatient family medicine clinic. In the hospital, she received a 14-day course of intravenous vancomycin for methicillin-resistant Staphylococcus aureus (MRSA) pneumonia. Her fingers became progressively painful. Nail bed changes were suggestive of infarction (Figure 4). She was started on warfarin to prevent further infarction and eventual ischemic necrosis. There was concern for possible antiphospholipid syndrome (APS). She was prescribed a short course of combination acetaminophen, 325 mg, and oxycodone, 7.5 mg, every 6 hours as needed for severe finger pain.

Outcome of the case. It remained unclear 4 months after initial presentation whether APS was a primary concern or was related to underlying MRSA pneumonia. There were no other signs of connective tissue disease, and the pain and discoloration of her fingers had completely resolved.
Laboratory tests revealed the following values: ANA, positive; antiphospholipid IgM antibodies, 18.8 MPL; and antiphospholipid IgG antibodies, 52.8 GPL. The medical team agreed that the patient should take warfarin indefinitely.
Rheumatology follow-up at 1 year confirmed RP, but clinically, the patient did not meet the American College of Rheumatology criteria for systemic lupus erythematosus (SLE). It still was uncertain whether the patient had infection-related APS or secondary RP associated with SLE. Connective tissue disease was ill-defined clinically with RP, APS, and multifactorial fatigue still features of her disease state.
Laboratory test results continued to point to a diagnosis of SLE. She eventually began a trial of hydroxychloroquine, 200 mg twice daily, for systemic symptoms including stiffness and discomfort in her joints, especially her hands. Warfarin was continued and titrated to a therapeutic international normalized ratio between 2 and 3.
The patient was lost to follow-up for several years due to changes in her living arrangements and insurance coverage. During this time, she had become supratherapeutic on warfarin therapy while taking antibiotics for an oral abscess. Her warfarin therapy was stopped and was never appropriately restarted. Another episode of RP occurred.
Despite the addition of enoxaparin and restarting warfarin, her right index finger required partial amputation due to wet gangrene. Eventually, she did follow up with her original care team. Warfarin was continued indefinitely, and she began further SLE-targeted treatment. In the 2 years that followed, she had no additional clinically significant episodes of RP, and her overall health began to improve.
Discussion. RP occurs in approximately 15% to 30% of patients with SLE.1 Such cases are classified as secondary RP. SLE is a heterogeneous autoimmune disease characterized by the presence of autoantibodies against nuclear antigens. It is a multisystem disease, and patients present in vastly different ways.2 SLE is more common in women, with prevalence rates ranging from 164 per 100,000 for white women to 406 per 100,000 for African American women.3 Probable causative factors include genetic predisposition, complement deficiencies, persistence of antigen, drugs, and environmental factors.4
The clinical course of SLE is incredibly variable and is characterized by acute and chronic episodes. The integumentary system is most often involved, and common manifestations include rash, arthritis, and fatigue.
Presentations associated with vasculitis include RP, pleuritis, and APS. In fact, vasculitis and RP are the most common vascular manifestations of SLE after systemic hypertension.5 Rare cases of digital gangrene in SLE patients have also been reported.6,7
Peripheral vascular disease also is common in SLE patients and is defined clinically as 1 or more episodes of intermittent claudication, absent or unequal pulses, gangrene, or ischemic ulcers, and subclinically as asymptomatic patients with abnormalities on Doppler ultrasonography, with 50% or greater reduction in diameter considered hemodynamically significant.8 Intermittent claudication along with gangrene was documented in our patient, further supporting a diagnosis of SLE. In such patients, the risk of thrombosis remains elevated throughout the course of the disease.9
Digital ulcers and gangrene are common skin manifestations of connective tissue disorders, although they are relatively rare in SLE.10 The integumentary system is a major target of SLE, yet digital ulcers and RP, being uncommon, are not included in the diagnostic criteria for SLE. Nevertheless, RP is a significant risk factor for vascular thrombosis in SLE patients with positive antiphospholipid antibodies.11,12
The prevalence of thrombosis in SLE patients with positive antiphospholipid antibodies is 38.8%.11 The risk of thrombosis is higher in SLE than in other autoimmune diseases.9 Early treatment can prevent complications of SLE, thus it becomes important to diagnose SLE at an early stage.
REFERENCES:
- Kammer GM, Soter NA, Schur PH. Circulating immune complexes in patients with necrotizing vasculitis. Clin Immunol Immunopathol. 1980; 15(4):658-672.
- Manson JJ, Rahman A. Systemic lupus erythematosus. Orphanet J Rare Dis. 2006;1:6.
- Chakravarty EF, Bush TM, Manzi S, Clarke AE, Ward MM. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56(6):2092-2094.
- Von Feldt JM. Systemic lupus erythematosus: recognizing its various presentations. Postgrad Med. 1995;97(4):79-86.
- Ansari A, Larson PH, Bates HD. Vascular manifestations of systemic lupus erythematosus. Angiology. 1986;37(6):423-432.
- Vocks E, Welcker M, Ring J. Digital gangrene: a rare skin symptom in systemic lupus erythematosus. J Eur Acad Dermatol Venereol. 2000;14(5): 419-421.
- Ziaee V, Yeganeh MH, Moradinejad M-H. Peripheral gangrene: a rare presentation of systemic lupus erythematosus in a child. Am J Case Rep. 2013;14:337-340.
- Bhatt SP, Handa R, Gulati GS, et al. Peripheral vascular disease in systemic lupus erythematosus. Lupus. 2007;16(9):720-723.
- Romero-Díaz J, García-Sosa I, Sánchez-Guerrero J. Thrombosis in systemic lupus erythematosus and other autoimmune diseases of recent onset. J Rheumatol. 2009;36(1):68-75.
- Rosato E, Molinaro I, Pisarri S, Salsano F. Digital ulcers as an initial manifestation of systemic lupus erythematosus. Intern Med. 2011;50(7): 767-769.
- Choojitarom K, Verasertniyom O, Totemchokchyakarn K, Nantiruj K, Sumethkul V, Janwityanujit S. Lupus nephritis and Raynaud’s phenomenon are significant risk factors for vascular thrombosis in SLE patients with positive antiphospholipid antibodies. Clin Rheumatol. 2008;27(3):345-351.
- Montuori R, Riccardi C. Acrocyanosis, Raynaud’s phenomenon and digital gangrene in a case of systemic lupus erythematosus [in Italian]. Minerva Med. 1968;59(10):515-521.