
Acquired Factor VIII Inhibitor Deficiency in an Elderly Man
Acquired factor VIII inhibitor deficiency, also known as acquired hemophilia, is a rare hematological disorder in nonhemophiliacs that develops in response to the spontaneous production of circulating immunoglobulin G (IgG) autoantibodies or alloantibodies to factor VIII, an essential blood clotting factor. The disorder is typically characterized by spontaneous ecchymoses and soft tissue hematomas that can lead to significant bleeding, requiring immediate medical attention. We report a case of acquired hemophilia in an elderly black man who presented with the sudden onset of a hematoma on the chest. A discussion of the background of this rare disorder and its signs and symptoms, possible etiology, and treatment options is also provided.
Case Presentation
A 69-year-old black man was transferred from a nursing home to the emergency department following the sudden onset of a 7x3-cm mass on the left pectoral region. The mass had been present for 1 day, exhibited local warmth, and was nonfluctuant, mobile, and slightly tender to palpation. The patient had no pain, fever, weight loss, chest pain, or shortness of breath, and there was no history of physical abuse or of similar masses. The physical examination also revealed multiple purpuric spots on the chest wall, which ranged from 1 cm to 3 cm in diameter. Purpuric lesions and bilateral pitting edema were noted in the lower extremities.
The patient’s medical history was significant for schizophrenia, chronic obstructive pulmonary disease (COPD), seizure disorder, dementia, and previous surgery for a left hip replacement. He was not allergic to any medications and was taking the following agents for his medical illnesses: phenytoin, olanzapine, albuterol, and albuterol/ipratropium and ipratropium inhalers. The patient had no history of bleeding disorders, cancer, diabetes, coronary artery disease, autoimmune disease, lymphoproliferative disorders, or factor VIII transfusion.
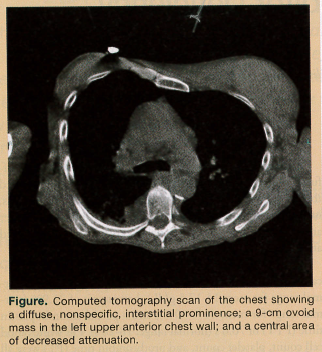 A computed tomography scan of the chest showed a diffuse, nonspecific, interstitial prominence, which reflected the chronic changes of COPD (Figure). A 9-cm ovoid mass, isodense with the musculature, was found in the left upper anterior chest wall. A questionable central area of decreased attenuation was seen, potentially indicating fluid collection.
A computed tomography scan of the chest showed a diffuse, nonspecific, interstitial prominence, which reflected the chronic changes of COPD (Figure). A 9-cm ovoid mass, isodense with the musculature, was found in the left upper anterior chest wall. A questionable central area of decreased attenuation was seen, potentially indicating fluid collection.
The biochemical profile was within normal limits. Significant laboratory values included a hemoglobin of 7.3 g/dL (normal range for men, 14-17.5 g/dL), hematocrit of 21.9% (normal range for men, 41%-50%), and a partial thromboplastin time (PTT) of 76.6 s (normal range, 18-28 s). His white blood cell count, platelet count, and prothrombin time (PT) were all within normal limits at 5300/µL (normal range, 4500-11000/µL), 179 x 103/µL (normal range, 150-350 x 103/µL), and 11.8 s (normal range, 10-13 s), respectively.
The patient was admitted to the medical teaching floor with an initial diagnosis of an abscess and was treated with vancomycin and piperacillin/tazobactam. He was given a transfusion of one unit of whole blood for low hemoglobin and hematocrit levels. None of the drugs he was taking were discontinued. Further evaluations included ultrasonography of the chest wall, and a gastroenterologist was consulted to rule out gastrointestinal (GI) bleeding and malignancy.
Over the next few days, the mass progressively increased in size toward the axilla. A thoracic surgeon was consulted and the hematoma was evacuated. During surgery, homeostasis was not achieved, and the patient continued to ooze blood from the muscle, although no active arterial bleeding was observed. The wound was packed with a hemostatic agent and dressing to promote clotting. Five thousand units of thrombin were applied and a Jackson-Pratt drain was placed before wound closure. The pathology report described fragments of blood clot with fibrin and fragments of soft tissue, including skeletal muscle with moderate to severe acute inflammation.
The patient continued to bleed from the wound site, and his hemoglobin and hematocrit levels continued to drop, requiring additional whole blood transfusions. At this point, a hematologist was consulted, and assays for various blood clotting factors were ordered.
The thoracic surgeon evaluated the patient again and repeated the evacuation of the hematoma at the bedside. The patient continued to bleed from the wound site, became hypotensive, and was transferred to the intensive care unit. Laboratory data revealed low factor VIII levels (<1%), indicating factor VIII inhibitor deficiency. Factors XI, II, IX, and X were all within normal limits, as were the results of the von Willebrand factor and lactate dehydrogenase assays. The patient was immediately started on oral prednisone 30 mg three times daily and intravenous (IV) gamma globulin 50 mg/kg daily.
The hematoma was evacuated again at the bedside for the third time. The clots were removed and the wound was packed tightly. The patient was hypotensive and hypoxemic during the procedure and was intubated. His blood pressure (BP) improved upon administration of IV fluids and a transfusion of packed red blood cells. Despite these efforts, the patient continued to bleed from the wound and was treated with three IV doses of 4800 µg of coagulation factor VIIa at 4 pm, 6 pm, and 9 pm, and was continuously monitored for hemostasis. A decrease in bleeding from the wound site was noted.
The next morning, the patient continued to bleed and have episodes of hypotension, with his BP ranging from 76/58 mm Hg to 112/67 mm Hg. On physical examination, edema of the extremities and genitalia were apparent, and congestion and bilaterally decreased breath sounds were heard on chest auscultation. A fourth dose of coagulation factor VIIa was started immediately along with gamma globulins. The patient continued to be hypotensive, with a BP of 64/34 mm Hg, and a dopamine and norepinephrine drip was initiated. The patient was lethargic, unresponsive, and continued to bleed from the chest wall. The healthcare proxy was consulted and a decision was made not to resuscitate. The patient died of cardiopulmonary arrest from hemorrhagic shock.
Discussion
The components of the coagulation cascade have been described in detail in many basic physiology texts. Briefly, coagulation takes place via intrinsic, extrinsic, and common pathways. Acquired factor VIII inhibitor deficiency is an immune-mediated disease characterized by the presence of autoantibodies or alloantibodies to human factor VIII, an essential component of the coagulation cascade. Whereas acquired factor VIII inhibitor deficiency is a frequent complication in severe hemophilia, it is relatively rare in nonhemophiliacs, with an incidence of 0.2 to 1 cases in 1 million patients reported annually.1
The antibodies in nonhemophiliac patients are typically circulating IgG autoantibodies, usually consisting of a mixture of heavy-chain subclasses of IgG4 and IgG1, with kappa or lambda light-chain specificity. These immunoglobulins attack factor VIII:C activity by binding to phospholipids, von Willebrand factor, or both, thereby inhibiting the procoagulation activity of factor VIII and its subsequent interaction with factor IX or factor X.
Fifty percent of acquired hemophilia cases have no underlying etiology.1 Of the remaining cases, conditions that have been associated with acquired factor VIII inhibitor deficiency include autoimmune disorders, such as rheumatoid arthritis and systemic lupus erythematosus; malignancies, both hematologic and solid-tumor; and skin diseases, such as psoriasis and pemphigus.2 Our patient did not have any of these diseases. Acquired factor VIII inhibitor deficiency has also been associated with allergic drug interactions to agents such as penicillins, sulfonamides, phenytoin, and some antimalarial drugs (5.6% of cases).1,3,4 Our patient was taking phenytoin for a seizure disorder. Whether this agent was related to his acquired factor VIII inhibitor deficiency was not determined, although it may have put him at risk.
Acquired factor VIII inhibitor deficiency in nonhemophiliac patients has also been seen in postpartum women, typically around 2 to 5 months after delivery. In rare circumstances, the inhibitor may develop during pregnancy. It is more commonly seen in the elderly, however, with a higher incidence noted in patients aged 60 to 80 years, with the median age of presentation between 60 and 67 years.2,5 Our patient’s age put him at increased risk for the disorder. The fact that the elderly often have comorbid conditions, as was the case with our patient, may explain why factor VIII inhibitor deficiency has been reported to have a mortality rate of more than 20%.1
Differential Diagnosis
Factor VIII inhibitors are associated with severe bleeding episodes in almost 90% of patients.1 The management of such hemorrhages can be challenging because the bleeding may be life-threatening, refractory to local treatment, or respond poorly to conventional clotting factor concentrates. According to a cohort study (n=172) conducted in the United Kingdom, acquired factor VIII inhibitor deficiency most frequently presents with subcutaneous, muscular, and GI hemorrhaging; however, retroperitoneal, genitourinary, and intracranial bleeding has also been seen.6 Our patient’s presentation was consistent with subcutaneous and muscular bleeding. Acquired factor VIII inhibitor deficiency can be distinguished from congenital factor VIII deficiency in that the latter deficiency is more typically characterized by hemarthoses.7 Other presentations of acquired factor VIII inhibitor deficiency include melena, hematuria, and iatrogenic bleeding.
Acquired factor VIII inhibitor deficiency should be considered in nonhemophiliac patients presenting with spontaneous hematomas and sudden life-threatening bleeding. Initial coagulation studies will typically show a prolonged activated PTT (aPTT), with normal PT, thrombin time, and platelet count8; however, the finding of isolated aPTT prolongation is nonspecific. Other conditions, such as disseminated intravascular coagulation; decreases in factors VIII, IX, XI, XII, high-molecular–weight kininogen, or prekallikrein; or the presence of anticoagulants, such as heparin and lupus anticoagulant or other clotting factor inhibitors, can all cause a prolonged aPTT.5,9 Of these causes, lupus anticoagulant is the most common.10
A mixing study should be performed to differentiate acquired hemophilia from other disease states. In the case of inhibitors, aPTT mixing studies using equal volumes of a patient’s plasma with normal plasma will not correct the aPTT. If clotting factor deficiencies are responsible, the aPTT will typically correct to within 4 seconds of the aPTT of normal plasma.8 Weak antibodies may not prolong the aPTT unless the mixture is incubated for at least 1 hour at 37ºC.11
Further testing is required to rule out heparin and lupus anticoagulant as potential causes. Heparin can be identified by taking a patient’s medical history, and its presence is suggested by a prolonged thrombin time in association with a normal reptilase time.12 Lupus anticoagulant is suggested when aPTT values during the mixing study are similar when taken at 0°C and after intubation at 37°C.12 Specific tests, such as the dilute Russell’s viper venom time test and the kaolin clotting time test, can be used to confirm the presence of lupus anticoagulants.13 A newly developed enzyme-linked immunosorbent assay appears to discriminate mild, moderate, or severe factor VIII antibodies from other circulating inhibitors, such as lupus anticoagulants.14
A Bethesda assay can be performed to quantify the factor VIII activity. One Bethesda unit (BU) is defined as the quantity of inhibitor that neutralizes 50% of factor VIII in normal plasma after incubation at 37ºC for 2 hours. The treatment protocol will vary depending on the factor activity levels. Acquired factor VIII inhibitors in nonhemophilia exhibit a complex nonlinear behavior, and as such, the titer may belie the seriousness of the illness.15 In cases such as that of our patient, the need to establish hemostasis takes priority over performing sophisticated diagnostic studies. Given the rapidly deteriorating status of our patient and the fact that the treatment consisted of recombinant activated factor VII, the Bethesda assay was not performed.
Approaches to Treatment
The aim of acquired hemophilia therapy is twofold: (1) to control the bleeding; and (2) to eliminate the inhibitor. Various treatment options have been shown to be effective, and the treatment that is administered should be assessed on a case-by-case basis.
Treating the Bleeding. For patients with low inhibitor titer (<5 BU), hemostasis can be achieved if the factor VIII levels can be brought to 30% to 50%. This can be accomplished using either desmopressin acetate (DDAVP) or infusions of factor VIII. DDAVP has been shown to be effective in patients with very low inhibitor titers (<3 BU), but is typically insufficient to provide hemostatis.5 DDAVP was not a good treatment option for our patient, as his rapidly deteriorating condition necessitated more aggressive forms of treatment. Infusions of human factor VIII may provide hemostasis. While no guidelines exist with respect to dosing,16 some authors recommend a dose of factor VIII, 200 mg/kg by IV bolus every 8 to 12 hours.17 Some clinicians double or triple the dosage given to patients with congenital factor VIII deficiency.18 Although porcine factor VIII replacement has been used as effective treatment in the past, it is no longer commercially available.5
In cases where the inhibitor titer is high (>5 BU), it is recommended that factor VIII bypassing agents, such as recombinant activated factor VII or activated prothrombin complex concentrate (aPCC) should be used. No studies have compared the relative efficacies of these two treatments. Extracorporeal immunoadsorption and plasmapheresis should be considered in cases with severe hemorrhaging and in those who do not respond to the aforementioned treatments. Recombinant activated factor VII enables thrombin to be created through the extrinsic pathway, thereby obviating the need for factor VIII. Studies have shown a 90% positive response rate.19 Recommended dosing is 90 mg/kg to 120 mg/kg every 2 to 3 hours by IV bolus until the bleeding has stopped.5 This dosage should be increased to 120 mg/kg to 270 mg/kg by IV bolus every 2.5 to 3 hours if no response is seen after two doses.19 Patients who do not respond within 24 hours are unlikely to do so with continued treatment. Although factor VIIa therapy was given to our patient, it only temporarily decreased the bleeding.
The only aPCC currently available in the United States is factor eight inhibitor bypass activity (FEIBA).5 A retrospective study of 34 patients with acquired hemophilia A showed an overall complete response rate of 86% with a dosing regimen of FEIBA at 75 units/kg every 8 to 12 hours and a median number of 10 doses to control a severe bleed.20
Eradication of the Inhibitor. Elimination of the inhibitor should be attempted using immunosuppression once hemostasis has been established.21 First-line therapy of methylprednisolone at a dose of 1 mg/kg daily has been shown to be 60% to 70% effective when used alone and 70% to 80% effective when combined with oral cyclophosphamide, 50 mg to 150 mg daily.21 Rituximab, cyclosporine, and immunoglobulins have all been reported to be effective second-line therapies.21 Steroid and immunoglobulin therapy was attempted for our patient, but was ineffective.
Conclusion
Our case report illustrates the devastating outcome that can occur from acquired factor VIII inhibitor deficiency. Although it is a relatively rare disorder, clinical suspicion should be raised in the setting of spontaneous bleeding into soft tissues, when the patient is older than 60 years, and when the patient is taking several medications simultaneously. A careful medication review is especially important because adverse reactions to certain drugs have been implicated in the development of acquired hemophilia.22 In our case, for example, the patient had been taking phenytoin, a medication associated with acquired hemophilia, for a seizure disorder. Whether phenytoin caused acquired hemophilia in our patient could not be determined. Sophisticated studies not commonly available in most hospitals would have been necessary to definitively show that relationship. Nevertheless, our case highlights the importance of assessing an older patient’s current and recently discontinued medications in the context of his or her medical history, clinical examination findings, and hematologic laboratory results. Therapy should be directed at achieving hemostasis and eradicating the factor VIII inhibitor through the use of immunomodulating therapies.
The authors report no relevant financial relationships.
References
1. Franchini M, Gandini G, Di Paolantonio T, Mariani G. Acquired hemophilia A: a concise review. Am J Hematol. 2005;80(1):55-63.
2. Hay CR. Acquired haemophilia. Baillieres Clin Haematol. 1998;11(2):287-303.
3. Shetty S, Bhave M, Ghosh K. Acquired hemophilia A: diagnosis, aetiology, clinical spectrum and treatment options. Autoimmun Rev. 2011;10(6):311-316.
4. Green D, Blanc J, Foiles N. Spontaneous inhibitors of factor VIII: kinetics of inactivation of human and porcine factor VIII. J Lab Clin Med. 1999;133(3):260-264.
5. Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Am Soc Hematol Educ Prog Book. 2006;2006(1):432-437.
6. Collins PW, Hirsch S, Baglin TP, et al; UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007;109(5):1870-1877.
7. Kunz JS, Williams PJ. Acquired factor VIII deficiency treated with porcine factor VIII infusion followed by cyclophosphamide. Hosp Physician. 2005;41(5):44-47. www.turner-white.com/memberfile.php?PubCode=hp_may05_factor.pdf. Accessed March 12, 2012.
8. Cohen AJ, Kessler CM. Acquired inhibitors. Baillieres Clin Haematol. 1996;9(2):331-354.
9. Boggio LN, Green D. Acquired hemophilia. Rev Clin Exp Hematol. 2001;5(4):389-404.
10. Chng WJ, Sum C, Kuperan P. Causes of isolated prolonged activated partial thromboplastin time in an acute care general hospital. Singapore Med J. 2005; 46(9):450-456.
11. Okoli OC, Kenechukwu NE, Orjioke N, Ekekwe I, Iroegbu N. Acquired factor VIII inhibitor disorder in a 95-year-old-woman. Resident & Staff Physician. 2007;53(7). www.hcplive.com/publications/Resident-and-Staff/2007/2007-07/2007-07_06. Accessed March 12, 2012.
12. Franchini M. Acquired hemophilia A. Hematology. 2006;11(2):119-125.
13. Kazmi MA, Pickering W, Smith MP, Holland LJ, Savidge GF. Acquired haemophilia A: errors in the diagnosis. Blood Coagul Fibrinolysis. 1998;9(7):623-628.
14. Sahud M. Laboratory diagnosis of inhibitors. Semin Thromb Hemost. 2000;26(2):195-203. www.thieme-connect.de/ejournals/abstract/sth/doi/10.1055/s-2000-9823. Accessed March 26, 2012.
15. Kessler CM. Acquired Hemophilia. 2nd ed. Princeton, NJ: Excerpta Medica; 1995:9-24.
16. Collins PW. Management of acquired haemophilia A—more questions than answers. Blood Coagul Fibrinolysis. 2003;14(suppl 1):S23-S27.
17. Kessler CM. New perspectives in hemophilia treatment. Am Soc Hematol Educ Prog Book. 2005;2005(1):429-435.
18. Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol. 2003;121(1):21-35.
19. Baudo F, de Cataldo F, Gaidano G; Italian registry of acquired hemophilia. Treatment of acquired factor VIII inhibitor with recombinant activated factor VIIa: data from the Italian registry of acquired hemophilia. Haematologica. 2004;89(6):759-761.
20. Sallah S. Treatment of acquired haemophilia with factor eight inhibitor bypassing activity. Haemophilia. 2004;10(2):169-173.
21. Hay CR, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors Organisation. Br J Haematol. 2006;133(6):591-605.
22. Giangrande P. Acquired hemophilia (monograph). Montreal, Quebec, Canada: World Federation of Hemophilia, 2005:3. www.wfh.org/2/docs/Publications/Diagnosis_and_Treatment/TOH38_Acquired_Hemophilia.pdf. Accessed March 27, 2012.