
Peer Reviewed
Infantile Systemic Hyalinosis
Authors:
Joy Goldstein, MD
Division of General Pediatrics, Children’s Hospital Los Angeles, California
Lauren Meepos, MD
Division of General Pediatrics, Children’s Hospital Los Angeles, California
Ronen Zipkin, MD
Division of Hospital Medicine, Children’s Hospital Los Angeles, California
Citation:
Goldstein J, Meepos L, Zipkin R. Infantile systemic hyalinosis. Consultant. 2019;59(3):90-92.
A 4-month-old girl presented with signs of joint pain and hyperpigmentation of the skin over the joints. She had been born at 33 weeks of gestation via cesarean delivery due to fetal distress to a mother with a history of systemic lupus erythematosus (SLE). The pregnancy had been complicated by a maternal SLE flare requiring hydroxychloroquine treatment during the second and third trimester and by placental insufficiency leading to intrauterine growth restriction (IUGR).
The girl had remained in the neonatal intensive care unit (NICU) for 5 weeks with a relatively uncomplicated course, primarily working on feeding and growing. One week prior to discharge from the NICU, at 4 weeks of age, she was noted to have reduced movement at the shoulders, and raising her arms appeared to cause her pain. She received a clinical diagnosis of Erb palsy and was discharged home with outpatient physical therapy.
After returning home, her signs of joint pain progressively worsened. She was unable to lift her arms at the shoulders and cried out with movement. At approximately 6 weeks of age, her parents noticed discoloration on the infant’s knuckles and ankles. She was admitted at an outside hospital for 5 days, during which the results of an extensive workup were unremarkable. She had normal levels of electrolytes, including phosphorus and calcium, as well as normal rheumatologic laboratory test results. Echocardiography findings were unremarkable. A radiologic skeletal survey showed periosteal reactions in both femurs and diffuse osteopenia but no fractures. She was discharged home without a diagnosis.
After discharge, her joint pain and skin discoloration continued to worsen. The joint pain and stiffness began to involve her elbows and knees, and the skin discoloration spread to her wrists and elbows. Her only other illness was an uncomplicated upper respiratory tract infection. She had never traveled outside of the state, and her parents denied any trauma.
Both parents were originally from Mexico and reported distant consanguinity at the level of a great-great-great grandparent. A maternal cousin had died at 2 years of age from an unknown bone disease.
PHYSICAL EXAMINATION
On examination, the patient was small for her age but was well-appearing. She had reduced movement of her extremities and cried with any manipulation of the shoulders, elbows, wrists, knees, and ankles. No obvious joint swelling or erythema were present. It was difficult to fully extend at the shoulder, elbow, and knee, although slow passive range of motion was possible. Symmetric, brownish purple hyperpigmentation was present bilaterally over the metacarpophalangeal (MCP) joints, the lateral aspect of the wrists, the medial aspect of the elbows, the medial and lateral malleoli, and the proximal interphalangeal joints of the feet (Figures 1 and 2).
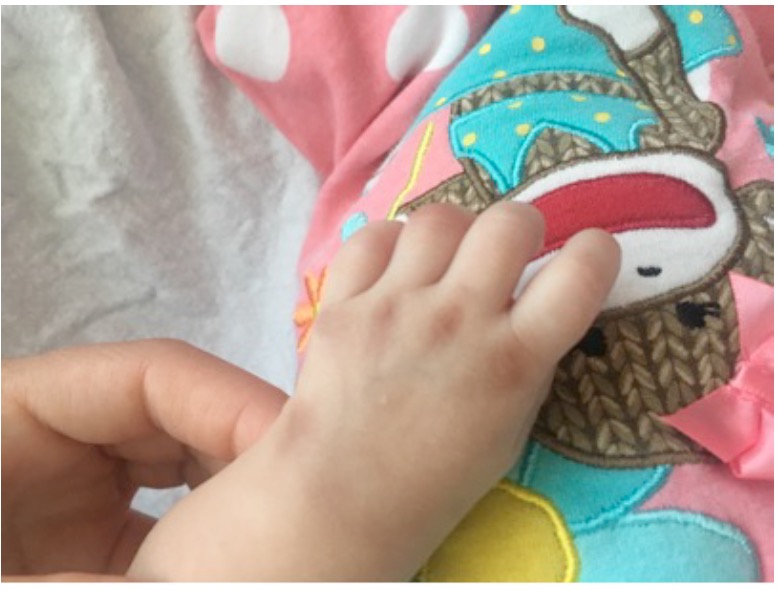
Figure 1. Hyperpigmentation over the patient’s MCP joints.
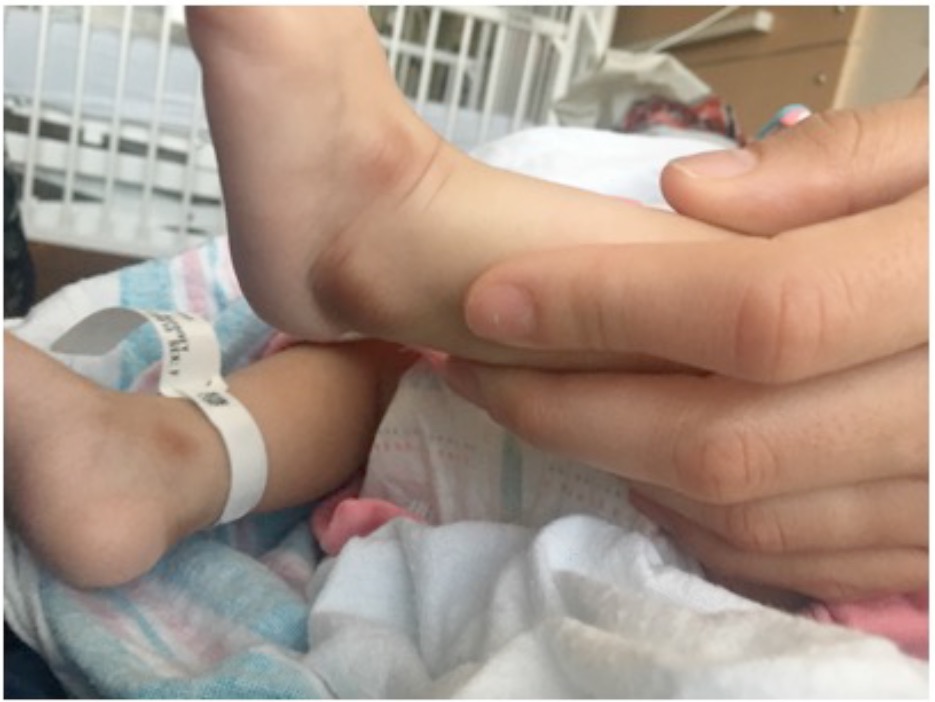
Figure 2. Hyperpigmentation over the patient’s medial and lateral malleoli.
A skeletal survey was performed, the results of which showed generalized slightly decreased bone density but no acute or healing fractures. Abdominal ultrasonography findings were normal. A complete blood count, comprehensive metabolic panel, C-reactive protein level, and erythrocyte sedimentation rate were obtained, the results of which were unremarkable. The 25-hydroxyvitamin D level was low at 12 ng/mL. The free thyroxine level was normal at 1.95 ng/dL, and the thyrotropin level was mildly elevated at 8.84 mIU/L.
She continued to appear to have significant pain with any manipulation of her extremities, which improved with the administration of morphine and ibuprofen.
Dermatology and genetics specialists were consulted, with further testing confirming the diagnosis of infantile systemic hyalinosis (ISH).
DISCUSSION
ISH is a rare autosomal recessive disease that involves deposition of hyaline material in multiple tissues throughout the body, resulting in joint contractures, subcutaneous nodules, diarrhea, and growth failure.1,2 It initially was described as a distinct disease in 1986 by Landing and Nadorra and is now considered to be a severe form of hyaline fibromatosis syndrome (HFS), which also includes juvenile hyaline fibromatosis, a similar condition that tends to be more mild.3,4
EPIDEMIOLOGY
ISH typically presents between birth and 6 months of age, with some cases presenting earlier with IUGR.1,2 It is equally common in males and females. While described in many ethnic groups, it has most commonly been seen in children of Turkish, Indian, and Moroccan descent.1
PATHOGENESIS
ISH is an autosomal recessive disorder caused by homozygous or compound heterozygous mutations in the anthrax toxin receptor gene (ANTXR2), also called the capillary morphogenesis protein 2 gene (CMG2), located on chromosome 4, long arm, band 21. This gene encodes a transmembrane protein that is upregulated in endothelial cells and may be involved in assembling basement membrane matrix.5 Patients with HFS have depositions of hyaline material in multiple tissues including skin, muscle, gastrointestinal tract, lymph nodes, spleen, thyroid, and adrenal glands.1 While the direct pathogenesis is not yet clear, it is possible that lack of the CMG2 protein leads to leakage of plasma components through the basement membrane into the perivascular space, resulting in collections of hyaline material.5,6 Alternatively, this protein may mediate the transport of type VI collagen to lysosomes for degradation, resulting in buildup of type VI collagen in the extracellular matrix in its absence.1 It appears that the location and type of mutation may determine disease severity within the spectrum of HFS.4
HISTOPATHOLOGY
Histopathology examination of skin lesions in ISH typically shows a normal epidermis and a dermis with deposits of eosinophilic and periodic acid–Schiff-positive substance in the extracellular and perivascular space, with proliferation of spindle cells without atypica.1
CLINICAL MANIFESTATIONS
The clinical manifestations of ISH can be categorized into osteoarticular findings, skin and mucosal findings, and systemic manifestations. Cognitive impairment is rare, likely because the CMG2 protein is minimally or not expressed in the brain.1
The first sign of ISH in a young infant may be extreme pain with minimal handling.1 Another early finding is joint involvement, including joint stiffness and pain, as well as contractures that ultimately result in frog-leg positioning and limited mobility.1,2,7 Patients are also at risk for osteopenia, osteoporosis, fractures, and osteolytic lesions of the long bones.7
A potential early skin finding that may help distinguish ISH involves hyperpigmented macules over bony prominences and joints, such as elbows, malleoli of the ankles, and MCP joints,8 as seen in our patient. Subsequent skin findings include pearly papules and subcutaneous nodules, most commonly on the head and in the perianal region, as well as generalized skin thickening.2,8 The skin lesions can be disfiguring but are otherwise asymptomatic. Patients often develop gingival hyperplasia that can be severe enough to interfere with feeding.7
Patients can develop intractable diarrhea with protein-losing enteropathy (PLE) secondary to deposition of hyaline material in the gastrointestinal tract, which can lead to failure to thrive.1,9 Hyaline material may also deposit in the heart, trachea, spleen, adrenal glands, skeletal muscle, thyroid, pancreas, lung, liver, and thymus and lymph nodes, although patients may not have obvious clinical manifestations.3,7,10
DIAGNOSIS
Early diagnosis requires clinical suspicion and is important to reduce hospital stay and unhelpful and potentially painful workup and treatments.3 The diagnosis can be confirmed with genetic testing for ANTXR2, with both homozygous and heterozygous variants seen in ISH. Positive genetic testing along with typical clinical manifestations confirm the diagnosis.1
DIFFERENTIAL DIAGNOSIS
In patients presenting with characteristic skin and joint findings, the differential diagnosis includes Winchester syndrome, lipoid proteinosis (Urbach-Wiethe disease), mucopolysaccharidosis type II (Hunter syndrome), Farber lipogranulomatosis, congenital generalized myofibromatosis, infantile stiff skin syndrome, and neonatal onset multisystemic inflammatory disease (NOMID) (Table).1,2,11,12
|
Table. Differential Diagnosis2,12-15 |
||||||
|
|
Inheritance |
Age at Onset |
Dermatologic Manifestations |
Clinical Features |
Survival |
Pathology |
|
Infantile systemic hyalinosis |
Autosomal recessive |
Infancy |
Diffusely thickened skin, small nodular thickenings, gingival hypertrophy |
Joint contractures, osteopenia, growth failure, diarrhea, frequent infections |
Up to 2 years |
Hyaline depositions |
|
Juvenile hyaline fibromatosis |
Autosomal recessive |
Childhood |
Nodular skin lesions, gingival hypertrophy |
Joint contractures, osteopenia |
Adulthood |
Hyaline depositions |
|
Winchester syndrome |
Autosomal recessive |
Infancy |
Patches of thickened leathery skin, coarse facies, gingival hypertrophy |
Short stature, severe joint contractures, peripheral corneal opacities, dissolution of carpal and tarsal bones, osteoporosis |
Adulthood |
Proliferation of fibroblasts in reticular dermis, followed by collagen appearing homogenized with few fibroblasts |
|
Lipoid proteinosis (Urbach-Wiethe disease) |
Autosomal recessive |
Birth to first few years of life |
Vesicular pustules that crust and heal with “icepick” acneiform scars; later yellowish, waxy papules, nodules, or plaques; beaded papules on eyelid margins |
Hoarse voice, intracranial calcifications |
Adulthood |
Hyaline depositions |
|
Mucopolysaccharidosis type II (Hunter syndrome) |
X-linked recessive |
First 2 years |
Generalized skin thickening, ivory-colored papules in scapular area and other sites of trauma, hypertrichosis, coarse facies |
Dysostosis with dwarfism, hepatosplenomegaly, cardiovascular disorders, deafness |
Severe, 15 years; mild, adulthood |
Metachromatic granules within fibroblasts and extracellular deposits between collagen bundles and fibers |
|
Farber lipogranulomatosis |
Autosomal recessive |
Early infancy |
Erythematous papules and subcutaneous nodules over joints, tendons, and pressure points |
Hoarseness, joint deformities, internal organs affected |
Infancy or childhood |
Accumulation of sphingolipid material |
|
Congenital generalized myofibromatosis |
Most sporadic |
Usually before age 2 |
Nodules in skin and subcutis |
Nodules in muscle, bone, and internal organs |
Variable, dependent on location of lesions |
Hyperproliferation of myofibroblasts |
|
Infantile stiff skin syndrome |
Autosomal dominant |
Infancy |
Stony hard skin, mild hypertrichosis |
Limited joint mobility, thoracic wall abnormalities |
Adulthood |
Fascial sclerosis or increased fibroblast cellularity with sclerotic collagen bundles in the deep reticular dermis |
|
Neonatal onset multisystemic inflammatory disease (NOMID) |
Most sporadic |
Infancy |
Widespread urticarial papules and plaques |
Short stature, arthropathy, chronic aseptic meningitis, periodic fever |
Adulthood |
Neutrophilic infiltration |
COMPLICATIONS
Patients with ISH often develop failure to thrive with chronic malnutrition, likely secondary to intractable diarrhea with malabsorption and PLE; severe gingival hyperplasia can lead to poor feeding, as well.1,3,7,12 They are also at risk for recurrent severe infections, which may in part be related to PLE.12
PROGNOSIS
ISH is a progressive disease that typically results in death by 2 years of age, most often secondary to intractable diarrhea, recurrent infections, and organ failure.1,4 Cases of longer survival have been reported.16,17
MANAGEMENT
Treatment of patients with ISH primarily involves supportive care, including pain management with nonsteroidal anti-inflammatory medications and opiates, and physical therapy as tolerated.1 Oral penicillamine has reportedly improved joint mobility in a few cases but overall has had limited success9,18; other medications including methotrexate and corticosteroids have been tried without significant benefit.18 Surgical excision of skin lesions can be considered, although recurrence can occur, and partial or radical gingivectomy may be helpful for gingival hyperplasia.4 For patients who develop failure to thrive, supplemental feeds via a nasogastric or gastrostomy tube should be considered.1
OUTCOME OF THE CASE
A dermatologist performed a punch biopsy of a hyperpigmented lesion on the infant’s left foot, the results of which demonstrated sclerosis of the deep dermis with elevation of the eccrine coils, with apparent loss of the peri-eccrine fat, and with relative loss of the CD34+ dermal dendritic cells at the base of the specimen. Genetic test results were positive for a homozygous mutation of ANTXR2, confirming the diagnosis of ISH. She was discharged home with oral morphine as needed for pain, physical therapy, and outpatient follow-up with medical genetics, palliative care, and dermatology specialists.
- Casas-Alba D, Martínez-Monseny A, Pino-Ramírez RM, et al. Hyaline fibromatosis syndrome: clinical update and phenotype-genotype correlations. Hum Mutat. 2018;39(12):1752-1763.
- Shin HT, Paller A, Hoganson G, Willner JP, Chang MW, Orlow SJ. Infantile systemic hyalinosis. J Am Acad Dermatol. 2004;50(2 suppl):S61-S64.
- Criado GR, González-Meneses A, Cañadas M, Rafel E, Yanes F, De Terreros IG. Infantile systemic hyalinosis: a clinicopathological study. Am J Med Genet A. 2004;129A(3):282-285.
- Denadai R, Raposo-Amaral CE, Bertola D, et al. Identification of 2 novel ANTXR2 mutations in patients with hyaline fibromatosis syndrome and proposal of a modified grading system. Am J Med Genet A. 2012;158A(4):732-742.
- Hanks S, Adams S, Douglas J, et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003;73(4)791-800.
- Dowling O, Difeo A, Ramirez MC, et al. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003;73(4):957-96
- Nofal A, Sanad M, Assaf M, et al. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: a unifying term and a proposed grading system. J Am Acad Dermatol. 2009;61(4):695-700.
- Stucki U, Spycher MA, Eich G, et al. Infantile systemic hyalinosis in siblings: clinical report, biochemical and ultrastructural findings, and review of the literature. Am J Med Genet. 2001 Apr 22;100(2):122-129.
- Lindvall LE, Kormeili T, Chen E, et al. Infantile systemic hyalinosis: case report and review of the literature. J Am Acad Dermatol. 2008;58(2):303-307.
- Glover MT, Lake BD, Atherton DJ. Infantile systemic hyalinosis: newly recognized disorder of collagen? Pediatrics. 1991;87(2)228-234.
- Aggarwal MLS, Chilakamarri V, Chennuri VS, Karra M. Identical twins with infantile systemic hyalinosis: case study and review of literature. J Orthop Case Rep. 2016;6(1):69-71.
- Urbina F, Sazunic I, Murray G. Infantile systemic hyalinosis or juvenile hyaline fibromatosis? Pediatr Dermatol. 2004;21(2):154-159.
- Larralde M, Ferrari B, Martinez JP, Barbieri MAF, Méndez JH, Casas J. Infantile myofibromatosis. An Bras Dermatol. 2017;92(6):854-857.
- Kilcline C, Shinkai K, Bree A, Modica R, Von Scheven E, Frieden IJ. Neonatal-onset multisystem inflammatory disorder: the emerging role of pyrin genes in autoinflammatory diseases. Arch Dermatol. 2005;141(2):248-253.
- Liu T, McCalmont TH, Frieden IJ, Williams ML, Connolly MK, Gilliam AE. The stiff skin syndrome: case series, differential diagnosis of the stiff skin phenotype, and review of the literature. Arch Dermatol. 2008;144(10):1351-1359.
- Thauvin-Robinet C, Faivre L, Beer F, Justrabo E, Nivelon-Chevallier A, Huer F. Infantile systemic hyalinosis: a case with atypical prolonged survival. Acta Paediatr. 2001;90(6):705-706.
- Al-Najjadah I, Bang RL, Ghoneim IE, Kanjoor JR. Infantile systemic hyalinosis. J Craniofac Surg. 2003;14(5):719-723.
- Al-Mayouf SM, AlMehaidib A, Bahabri S, Shabib S, Sakati N, Teebi AS. Infantile systemic hyalinosis: a fatal disorder commonly diagnosed among Arabs. Clin Exp Rheumatol. 2005;23(5):717-720.