
Peer Reviewed
Acute Pancreatitis: History, Scoring System, and Treatment
Authors:
Michael Krzyzak, MD
Department of Medicine, Staten Island University Hospital, Staten Island, New York
Stephen Mulrooney, MD
Department of Gastroenterology, Staten Island University Hospital, Staten Island, New York
Citation: Krzyzak M, Mulrooney S. Acute pancreatitis: history, scoring system, and treatment [published online September 12, 2018]. Gastroenterology Consultant.
Abstract: Acute pancreatitis is a frequent occurrence in hospital practice. Presenting with epigastric pain, it is diagnosed as fulfilling 2 of 3 criteria, including laboratory, clinical, and radiologic findings. Treatment is based on aggressive intravenous hydration of lactated Ringer solution and pain management. Enteral nutrition should be started as soon as it can be tolerated. Complications from acute pancreatitis include recurrent episodes, chronic pancreatitis, and necrosis.
The word pancreas originates from ancient Greek pan- + kreas, meaning “all flesh.”1 Herophilus of Chalcedon, noted as one of the founders of the ancient school of medicine in Alexandria, is credited as the first person to describe the pancreas, but the organ was named by Rufus of Ephesus.2 The name all flesh comes from its gross description as an organ without any bone or cartilage. By the 16th century, the pancreas had been described as ductal system into which duodenal chyme ascended.3 Pancreatitis was first described in 1652 by Nicolaes Tulp, a Dutch surgeon and lecturer of anatomy, who is remembered as the subject on Rembrandt’s “The Anatomy Lesson of Dr. Nicolaes Tulp” (Figure).4,5 Pancreatitis was further described as hemorrhagic, suppurative, and gangrenous in the 19th century. During this time, the clinical signs of epigastric pain were described as "deep-seated, dull, epigastric pain, distention, sickness, and vomiting."6

Figure. “The Anatomy Lesson of Dr. Nicolaes Tulp” (1632), by Rembrandt.5
A diagnosis of acute pancreatitis requires 2 of the following 3 criteria: abdominal pain, an amylase/lipase level 3 times the upper limit of normal, and findings on imaging that are suggestive of pancreatitis.7
The annual incidence of pancreatitis is reported to be from 13 to 45 per 100,000 US population.8 Inciting events for pancreatitis vary widely. In the pediatric population, trauma, abuse, viral infections, hemolytic-uremic syndrome, gallstones, Shwachman-Diamond syndrome, and cystic fibrosis are among the causes.4 The African American population has a 2- to 3-fold higher risk of developing acute pancreatitis than the white population, and men and women are equally affected.9
Initially, pancreatitis was thought to be attributable only to gallstones.10 In a prospective US study, 37.9% of pancreatitis cases were gallstone-related, 43.5% were not gallstone-related, and 18.6% were cases of recurrent acute pancreatitis.9 Gallstones and alcohol use are the top 2 culprits of pancreatitis.11 Smoking was found to have an association, with a hazard ratio of 2.01 reported in women with more than 20 pack-years of smoking.9 Less than 5% of cases of acute pancreatitis are linked to medication use, and although an extensive list of medications is associated with pancreatitis, it is difficult to determine whether a drug is responsible.12 Medications such as proton-pump inhibitors, antibacterials, azathioprine, enalapril, mesalamine, 6-mercaptopurine, and valproic acid are among the common medication-induced causes of pancreatitis.12,13
Chiari proposed the autodigestion of pancreatic enzymes in 1896.4 The agitator of the proinflammatory response can be physical, chemical, or both, such as in post–endoscopic retrograde cholangiopancreatography (ERCP) pancreatitis (PEP). Numerous inciting factors have been noted (Table).14-17
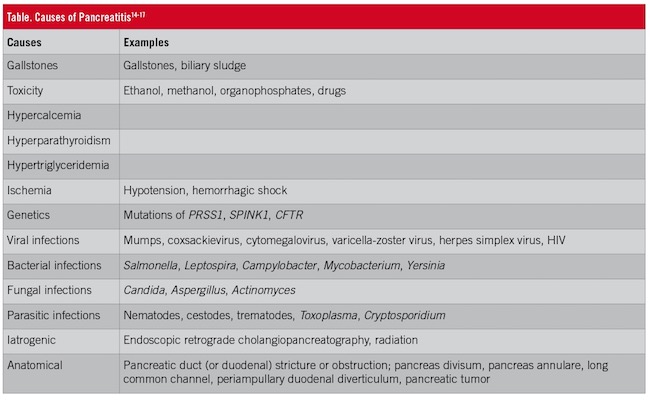
The inciting event can vary but must produce pancreatic duct obstruction, which impedes exocytosis of zymogen granules from acinar cells. Zymogen granules contain digestive enzymes that begin the process of autodigestion.18 Two hypotheses, the reflux hypothesis and colocalization hypothesis, have been developed to explain the initiation of the process. The reflux hypothesis relies on bile reflux via the pancreatic duct activating trypsinogen.19 Alternatively, the colocalization theory relies on lysosomal colocalization within the acinar cells. In cellular models, cathepsin B and N-acetylglucosaminidase become redistributed during pancreatic duct obstruction. These lysosomal hydrolases localize in zymogen granules in acinar cells.20
The acinar cells initiate the cascade via 1 of 4 proposed mechanisms: cathepsin B activation, disruption in calcium signaling, trypsinogen autoactivation, and inappropriate trypsinogen activation. Trypsinogen autoactivates in the acinar cells and progresses to macrophages, which also activate trypsinogen via cathepsin B. This increases the severity of the proinflammatory response. Interleukins have been well documented as part of the inflammatory cascade. Interleukin 6 (IL-6), IL-8, IL-1β, soluble IL-2 receptor (sIL-2R), and C-reactive protein (CRP) have been documented to correlate with the severity of pancreatitis.21-23 IL-1β and sIL-2R levels have been shown to elevate at day 1 of presentation, while IL-6, IL-8, and CRP levels have been found to be elevated at day 3.21 This amplification initiates a systemic response. The importance of the final pathway of inciting a proinflammatory response can be seen in the development of ameliorating strategies, such as in the use of rectal indomethacin with aggressive hydration.24
Acute Pancreatitis Scoring System
From the mid 20th century, monitoring patients’ clinical status, electrolytes, and blood cell count was recommended at regular intervals to guide treatment in acute pancreatitis.10
In 1977, Ranson and Pasternack narrowed down 43 variables to 11 variables to determine the severity of pancreatitis.25 These Ranson criteria included age, white blood cell count, and levels of blood glucose, serum lactate dehydrogenase, and serum aspartate transaminase, and 48 hours later, hematocrit decrease, blood urea nitrogen (BUN) increase, lowest serum calcium level, lowest arterial oxygen tension, highest base deficit, and estimated fluid sequestration.25
In 1989, the Acute Physiology and Chronic Health Enquiry, or APACHE-II, was applied as a scoring system to grade the severity of pancreatitis.26With 73% sensitivity and 84% specificity, APACHE-II can predict pancreatic necrosis between 0 and 48 hours of initial presentation. In comparison, the sensitivity and specificity of the Ranson criteria are 65% and 61% respectively.26
In 1990, Balthazar and colleagues presented a scoring system based on computed tomography (CT) scans.27 It utilized a 5-tiered scoring system from A to E depending on the degree of attenuation, which corresponds to progression of pancreatitis severity and fluid collection. The authors found that using the scoring system, the combined morbidity rate in patients with more than 30% necrosis was 94%, and the mortality rate was 29%.27 More importantly, the Balthazar scoring system provided an indicator of the development of a pancreatic abscess, with a positive predictive value of 84%.27
Six additional scoring systems have been developed: CT scoring systems; modified CT severity index; pancreatic size index; extrapancreatic score; extrapancreatic inflammation on CT score; and mesenteric edema and peritoneal fluid score, all of which take into account pancreatic, peripancreatic, and extrapancreatic features.28
In 2008, Wu and colleagues presented a new simplified method for assessing severity of pancreatitis using BUN, impaired mental status, systemic inflammatory response syndrome (SIRS), age greater than 60 years, and pleural effusion.29 Termed the bedside index for severity in acute pancreatitis (BISAP; the acronym also derives from each of the 5 criteria used), it provides an easy-to-use method of assessment that requires only a 1-time evaluation of laboratory test results compared with earlier criteria requiring evaluation at presentation and again 48 hours later. A BISAP score of 3 or greater predicts higher mortality among patients, with sensitivity of 71%, specificity of 83%, a positive predictive value of 17.5%, and a negative predictive value of 99% for mortality.30 A stepwise increase in mortality is found with each additional point on BISAP: 0 points, 0.1% mortality rate; 1 point, 0.4% mortality rate; 2 points, 1.6% mortality rate; 3 points, 3.6% mortality rate; 4 points, 7.4% mortality rate; and 5 points, 9.5% mortality rate.31
In 1992, at the International Symposium on Acute Pancreatitis, a system was devised to classify the severity of pancreatitis.32 In 2012, the classification system was revised. Using the system, acute pancreatitis can be subdivided into 2 groups: interstitial edematous pancreatitis and necrotizing pancreatitis. Severity is classified as mild, moderate, or severe. Mild pancreatitis does not have any systemic complications including organ failure. Moderate pancreatitis is defined by transient systemic complications. Severe acute pancreatitis is characterized by consistent organ failure.32
Other markers described in the literature include procalcitonin, antithrombin III, and CRP. Increases in levels of 1 of these markers correlate with an increase in mortality.33,34
Management of Acute Pancreatitis
Fluid resuscitation is essential to the treatment of acute pancreatitis. From 250 to 500 mL/h of isotonic crystalloid solution should be provided to all patients with acute pancreatitis unless cardiovascular, renal, or other related comorbid factors exist in the first 24 hours.7 Systemic inflammation causes third-spacing due to inflammation. Aggressive hydration is required for microcirculatory and macrocirculatory support. Increased perfusion by hydration prevents additional inflammation and necrosis.7
Current recommendations support lactated Ringer solution as the preferred choice of hydration.7 Fluid resuscitation with a pH-balanced solution as lactated Ringer was found to have decreased CRP levels and decreased SIRS incidence.35 A less-acidic solution leads to less acinar injury and a decreased systemic inflammatory process.
Antibiotics are not administered routinely in acute pancreatitis cases. In severe pancreatitis with necrosis, prophylactic antibiotics did not show benefit compared with placebo. No reduction in progression to infection has been reported with the use of prophylactic antibiotics. Antibiotics are reserved for cases in which infection is suspected.36-38
Animal studies showed promise in utilizing probiotics in the treatment of severe acute pancreatitis,39 but in application to humans in prospective clinical trials, mortality was noted to be increased.40 A meta-analysis of probiotic prophylaxis concluded that probiotic prophylaxis was neither beneficial nor harmful.41 Similarly, prophylactic antibiotics are not recommended unless signs of infection are present.7,37
Originally, treatment relied on a conservative approach whereby the pancreas was given ample time to rest from secretion of cholecystokinin and secretin.10,42 To do this, enteral feeding was given via nasogastric tube on the greater curvature of the stomach in order to prevent acid stimulation, at which point pH was checked regularly and antacids added as needed to prevent acid stimulation. A pH of 5 was considered optimal for treatment.42 Since then, much attention has been given to enteral or parenteral treatment.
Parenteral nutrition had been thought to avoid pancreatic stimulation and provide time to rest for the pancreas; however, studies suggest parenteral nutrition exacerbates a proinflammatory factor and cause immunosuppression. Treatment with parenteral nutrition also is more expensive and can lead to more septic complications. Thus it is recommended to begin enteral nutrition as soon as it can be tolerated.43,44 Furthermore, nutrition had been originally focused on bypassing the stomach to avoid gastric ileus.45 Numerous systematic reviews of studies have shown no advantage of nasogastric feeding over nasojejunal feeding.45,46 Patients with mild uncomplicated pancreatitis do not benefit from nutritional support and can follow a normal diet once it is tolerated. Nutritional support should begin early in the course of patients with moderate to severe disease.47
Other guidelines emphasize the need for abdominal ultrasonography to exclude gallstone etiology. If there is evidence of biliary obstruction or persistent obstruction of the common bile duct and pancreatic duct on ultrasonography, CT, or magnetic resonance cholangiopancreatography, consideration should be given to therapeutic ERCP.7
Complications
Complications of pancreatitis can occur early or late (>1 week) in the course of the disease. Local complications in the pancreas include peripancreatic fluid collections, necrosis, pseudocysts, and walled-off necrosis.7,32 Intervention is required for persistent areas of necrosis that are infected or symptomatic. Initially treated with open pancreatic necrosectomy, reports have shown mortality rates from 6% to 47%. New modalities focus on video-assisted retroperitoneal debridement or peroral necrosectomy.48-50
Between 17% and 20% of patients who have had an episode of severe pancreatitis can have a recurrent episode, and between 8% and 30% of patients with acute pancreatitis progress to chronic pancreatitis.51,52 Smoking and heavy alcohol use were found to be factors that increase recurrence of pancreatitis. Heavy drinking, defined as more than 5 drinks per day, was found to have an odds ratio of 3.10 of having recurrent pancreatitis.53
Conclusion
Multiple causes of acute pancreatitis have been described, including physical causes, chemical causes, or both. The pathologic mechanism is based on activation of trypsinogen, but further research is required to fully elaborate the inciting mechanism. A combination of epigastric pain, radiologic evidence, and laboratory findings help diagnose acute pancreatitis. Recent literature findings support early enteral nutrition, intravenous lactated Ringer solution, and pain management as the core of treatment. Severity can be graded using classification systems such as the BISAP scoring system. Complications from an episode of acute pancreatitis are recurrent episodes, pancreatic necrosis, and progression to chronic pancreatitis.
References:
- Pancreas. Merriam-Webster. https://www.merriam-webster.com/dictionary/pancreas. Updated July 13, 2018. Accessed July 20, 2018.
- Tsuchiya R, Fujisawa N. On the etymology of “pancreas.” Int J Pancreatol. 1997;21(3):269-272.
- Busnardo AC, DiDio LJA, Tidrick RT, Thomford NR. History of the pancreas. Am J Surg. 1983;146(5):539-550.
- Modlin IM, Champaneria MC, Chan AKC, Kidd M, Eick GN. The history of the pancreas. In: Beger HG, Warshaw AL, Büchler MW, et al, eds. The Pancreas: An Integrated Textbook of Basic Science, Medicine, and Surgery. 2nd ed. Malden, MA: Blackwell Publishing Ltd; 2008:9-41.
- IJpma FFA, van de Graaf RC, Nicolai J-PA, Meek MF. The anatomy lesson of Dr Nicolaes Tulp by Rembrandt (1632) and the findings during dissection of the forearm of a cadaver: anatomical discrepancies [in Dutch]. Ned Tijdschr Geneeskd. 2006;150(50):2756-2765.
- Fitz RH. Acute pancreatitis—a consideration of pancreatic hemorrhage, hemorrhagic, suppurative and gangrenous pancreatitis, and of disseminated fat-necrosis. Boston Med Surg J. 1889;70(8):181-187.
- Tenner S, Baillie J, DeWitt J, Vege SS. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol. 2013;108(9):1400-1415,1416.
- Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1252-1261.
- Setiawan VW, Pandol SJ, Porcel J, et al. Prospective study of alcohol drinking, smoking, and pancreatitis: the Multiethnic Cohort. Pancreas. 2016;45(6):819-825.
- Berk JE. The treatment of pancreatitis. Pa Med J. 1953;56(4):265-269.
- Yadav D, Hawes RH, Brand RE, et al; North American Pancreatic Study Group. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med. 2009;169(11):1035-1045.
- Forsmark CE, Vege SS, Wilcox CM. Acute pancreatitis. N Engl J Med. 2016;375(20):1972-1981.
- Bertilsson S, Kalaitzakis E. Acute pancreatitis and use of pancreatitis-associated drugs: a 10-year population-based cohort study. Pancreas. 2015;44(7):1096-1104.
- Campbell F, Verbeke CS. Pathology of the Pancreas: A Practical Approach. London, England: Springer-Verlag; 2013.
- Buse JB, Bethel MA, Green JB, et al; TECOS Study Group. Pancreatic safety of sitagliptin in the TECOS Study. Diabetes Care. 2017;40(2):164-170.
- Tkáč I, Raz I. Combined analysis of three large interventional trials with gliptins indicates increased incidence of acute pancreatitis in patients with type 2 diabetes. Diabetes Care. 2017;40(2):284-286.
- DeVries JH, Rosenstock J. DPP-4 inhibitor–related pancreatitis: rare but real! Diabetes Care. 2017;40(2):161-163.
- Lankisch PG, Apte M, Banks PA. Acute pancreatitis. Lancet. 2015;386(9988):85-96.
- Ferdek PE, Jakubowska MA, Gerasimenko JV, Gerasimenko OV, Petersen OH. Bile acids induce necrosis in pancreatic stellate cells dependent on calcium entry and sodium-driven bile uptake. J Physiol. 2016;594(21):6147-6164.
- Saluja A, Saluja M, Villa A, et al. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest. 1989;84(4):1260-1266.
- Mayer J, Rau B, Gansauge F, Beger HG. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut. 2000;47(4):546-552.
- Leser H-G, Gross V, Scheibenbogen C, et al. Elevation of serum interleukin-6 concentration precedes acute-phase response and reflects severity in acute pancreatitis. Gastroenterology. 1991;101(3):782-785.
- Gross V, Andreesen R, Leser H-G, et al. Interleukin-8 and neutrophil activation in acute pancreatitis. Eur J Clin Invest. 1992;22(3):200-203.
- Mok SRS, Ho HC, Shah P, Patel M, Gaughan JP, Elfant AB. Lactated Ringer’s solution in combination with rectal indomethacin for prevention of post-ERCP pancreatitis and readmission: a prospective randomized, double-blinded, placebo-controlled trial. Gastrointest Endosc. 2017;85(5):1005-1013.
- Ranson JHC, Pasternack BS. Statistical methods for quantifying the severity of clinical acute pancreatitis. J Surg Res. 1977;22(2):79-91.
- Larvin M, McMahon MJ. APACHE-II score for assessment and monitoring of acute pancreatitis. Lancet. 1989;2(8656):201-205.
- Balthazar EJ, Robinson DL, Megibow AJ, Ranson JH. Acute pancreatitis: value of CT in establishing prognosis. Radiology. 1990;174(2):331-336.
- Bollen TL, Singh VK, Maurer R, et al. A comparative evaluation of radiologic and clinical scoring systems in the early prediction of severity in acute pancreatitis. Am J Gastroenterol. 2012;107(4):612-619.
- Wu BU, Johannes RS, Sun X, Tabak Y, Conwell DL, Banks PA. The early prediction of mortality in acute pancreatitis: a large population-based study. Gut. 2008;57(12):1698-1703.
- Singh VK, Wu BU, Bollen TL, et al. A prospective evaluation of the bedside index for severity in acute pancreatitis score in assessing mortality and intermediate markers of severity in acute pancreatitis. Am J Gastroenterol. 2009;104(4):966-971.
- Layer P. A simple index on the day of admission which predicts outcome in acute pancreatitis. Gut. 2008;57(12):1645-1646.
- Bradley EL III. A clinically based classification system for acute pancreatitis: summary of the International Symposium on Acute Pancreatitis, Atlanta, Ga, September 11 through 13, 1992. Arch Surg. 1993;128(5):586-590.
- Bezmarević M, Kostić Z, Jovanović M, et al. Procalcitonin and BISAP score versus C-reactive protein and APACHE II score in early assessment of severity and outcome of acute pancreatitis. Vojnosanit Pregl. 2012;69(5):425-431.
- Donati V, Brogi E, Vetrugno L, Calamai I, Forfori F. Antithrombin: a possible new prognostic marker and therapeutic tool for acute pancreatitis? J Gastroenterol Dig Dis. 2016;1(2):23-24.
- Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):710-717.e1.
- Dellinger EP, Tellado JM, Soto NE, et al. Early antibiotic treatment for severe acute necrotizing pancreatitis: a randomized, double-blind, placebo-controlled study. Ann Surg. 2007;245(5):674-683.
- Isenmann R, Rünzi M, Kron M, et al; German Antibiotics in Severe Acute Pancreatitis Study Group. Prophylactic antibiotic treatment in patients with predicted severe acute pancreatitis: a placebo-controlled, double-blind trial. Gastroenterology. 2004;126(4):997-1004.
- Forsmark CE, Vege SS, Wilcox CM. Acute pancreatitis. N Engl J Med. 2017;376(6):598-599.
- van Minnen LP, Timmerman HM, Lutgendorff F, et al. Modification of intestinal flora with multispecies probiotics reduces bacterial translocation and improves clinical course in a rat model of acute pancreatitis. Surgery. 2007;141(4):470-480.
- Besselink MGH, van Santvoort HC, Buskens E, et al; Dutch Acute Pancreatitis Study Group. Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;371(9613):651-659.
- Gou S, Yang Z, Liu T, Wu H, Wang C. Use of probiotics in the treatment of severe acute pancreatitis: a systematic review and meta-analysis of randomized controlled trials. Crit Care. 2014;18(2):R57.
- Banks PA. Treatment of acute pancreatitis. In: Banks PA. Pancreatitis. New York, NY: Plenum Publishing Corp; 1979:109-129.
- Marik PE, Zaloga GP. Meta-analysis of parenteral nutrition versus enteral nutrition in patients with acute pancreatitis. BMJ. 2004;328(7453):1407.
- Kalfarentzos F, Kehagias J, Mead N, Kokkinis K, Gogos CA. Enteral nutrition is superior to parenteral nutrition in severe acute pancreatitis: results of a randomized prospective trial. Br J Surg. 1997;84(12):1665-1669.
- Petrov MS, Correia MITD, Windsor JA. Nasogastric tube feeding in predicted severe acute pancreatitis. A systematic review of the literature to determine safety and tolerance. JOP. 2008;9(4):440-448.
- Eatock FC, Chong P, Menezes N, et al. A randomized study of early nasogastric versus nasojejunal feeding in severe acute pancreatitis. Am J Gastroenterol. 2005;100(2):432-439.
- Pisters PW, Ranson JH. Nutritional support for acute pancreatitis. Surg Gynecol Obstet. 1992;175(3):275-284.
- Freeman ML, Werner J, van Santvoort HC, et al. Interventions for necrotizing pancreatitis: summary of a multidisciplinary consensus conference. Pancreas. 2012;41(8):1176-1194.
- Fernandez-del Castillo C, Warshaw AL. Parenchymal necrosis: infection and other indications for debridement and drainage [in German]. Chirurg. 2000;71(3):269-273.
- Reddy M, Jindal R, Gupta R, Yadav TD, Wig JD. Outcome after pancreatic necrosectomy: trends over 12 years at an Indian centre. ANZ J Surg. 2006;76(8):704-709.
- Ahmed Ali U, Issa Y, Hagenaars JC, et al; Dutch Pancreatitis Study Group. Risk of recurrent pancreatitis and progression to chronic pancreatitis after a first episode of acute pancreatitis. Clin Gastroenterol Hepatol. 2016;14(5):738-746.
- Cavestro GM, Leandro G, Di Leo M, et al. A single-centre prospective, cohort study of the natural history of acute pancreatitis. Dig Liver Dis. 2015;47(3):205-210.
- Durbec JP, Sarles H. Multicenter survey of the etiology of pancreatic diseases: relationship between the relative risk of developing chronic pancreatitis and alcohol, protein and lipid consumption. Digestion. 1978;18(5-6):337-350.