
Peer Reviewed
Hereditary Multiple Osteochondromas
AUTHORS:
Zackary M. Funk, BS1 • Priya G. Sharma, MD2 • Carolyn G. Carter, MD, MS3
AFFILIATIONS:
1University of Florida, College of Medicine, Gainesville, Florida
2Department of Radiology, University of Florida College of Medicine, Gainesville, Florida
3Department of Pediatrics, University of Florida College of Medicine, Gainesville, Florida
CITATION:
Funk ZM, Sharma PG, Carter CG. Hereditary multiple osteochondromas. Consultant. 2021;61(12):e42-e46. doi:10.25270/con.2021.04.00002
Received September 9, 2020. Accepted January 7, 2021. Published online April 7, 2021.
DISCLOSURES:
The authors report no relevant financial relationships.
CORRESPONDENCE:
Carolyn G. Carter, MD, 1600 SW Archer Road, PO Box 343, Gainesville, FL 32608 (cartcg@shands.ufl.edu)
A 9-month-old, full-term boy was brought to our pediatric clinic by his parents for his well-child check-up. The infant was noted to be afebrile and growing well, with a weight in the 52nd percentile and a length in the 95th percentile.
The patient had a history of a small right-sided pneumothorax, with little clinical significance at birth, that resolved after a few hours under a 100% oxyhood.
Physical examination. At the time of presentation to our clinic, 2 bony prominences were noted upon palpation of the lower left side of the ribcage, which prompted an anteroposterior chest radiography scan to be conducted (Figure 1). A review of systems and physical examination findings were otherwise normal. Family history was also not significant.
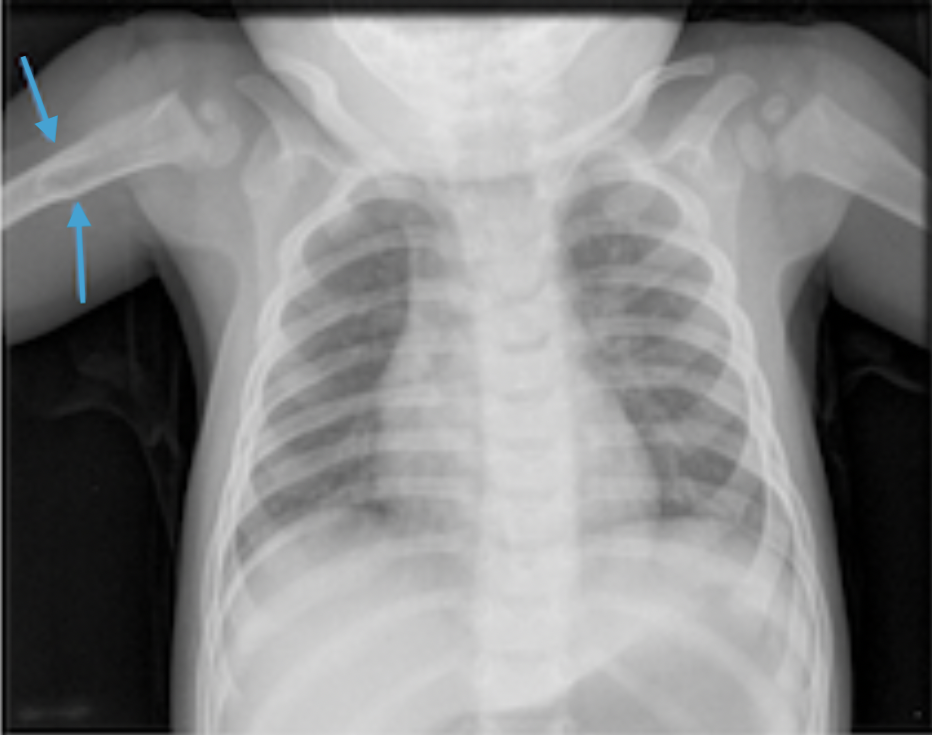
No bony abnormalities or other findings in the chest wall were visible on imaging. However, the radiologist did note a focal area of increased density in the right proximal humerus, which was thought to be secondary to artifact. This finding did not appear on the chest radiograph obtained at birth during an evaluation of the patient’s pneumothorax.
A few years later, when the patient was aged 2 years and 7 months, he and his mother presented to our clinic for another follow-up. The boy’s mother noted new palpable masses over his right shoulder, at which time additional plain diagnostic scans were conducted. At that time, the diagnosis of hereditary multiple osteochondromas (HMO), or multiple hereditary exostoses (MHE), was made.
The cortical abnormality thought to be artifact at his 9-month visit had since progressed and was now described as a well-corticated metadiaphyseal sessile lesion measuring 1.5 × 1.2 cm projecting away from the epiphysis, classic for the presentation of HMO (Figures 2a and 2b).
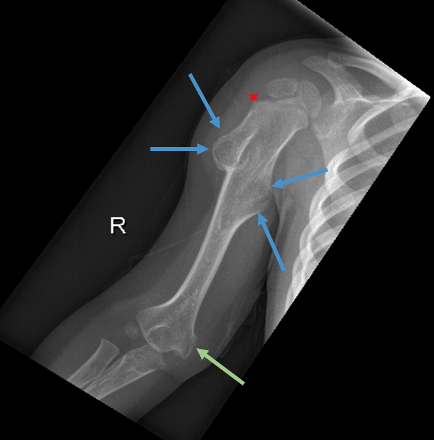
The diagnosis was supported radiographically upon further imaging of the lower extremities, which concluded that there were sessile and mildly pedunculated osteochondromas along the proximal and distal femurs (Figure 3), left tibia, and right fibula. Over time, the patient had developed multiple osteochondromas throughout his right upper extremity, left distal ulna, pelvis, bilateral femurs and tibia, right calcaneus, and left forefoot. At least 13 lesions were visualized in the radiographs (Figures 1-4).

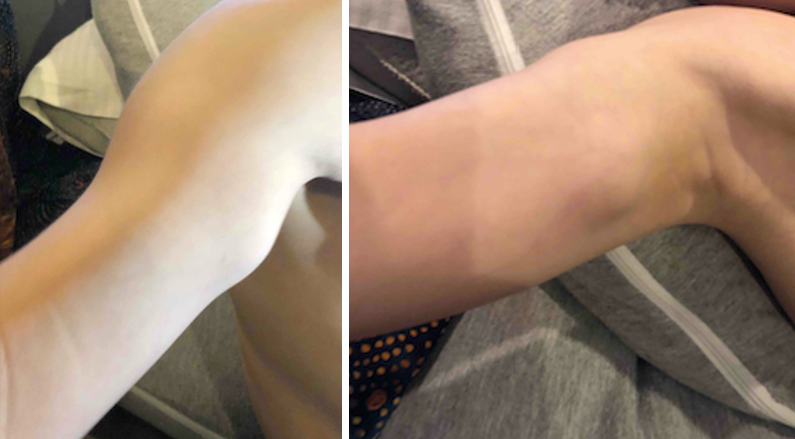
Two years later, when the patient was aged 4 years and 7 months, the carrying angle of his right upper extremity showed excess valgus positioning and relative shortening of the distal ulna (Figure 4). Although no intervention was necessary at that time, surgical management could eventually be indicated, including hemiepiphysiodesis of the distal radius or resection of osteochondromas to correct relative growth limitation of the distal ulna to avoid compression of the ulnar nerve by distal humeral osteochondroma.

Discussion. HMO, or MHE, is a condition linked to an autosomal dominant germline mutation in genes coding for exostosin glycosyltransferase (EXT) tumor suppressor genes, namely in EXT1 on chromosome 8 or EXT2 on chromosome 11.1,2 There has also been an association with mutations in EXT3 on chromosome 19 and in various genes in the exostosin-like (EXTL) family.2 Mutations in these genes are present in individuals with HMO and in patients with isolated chondrosarcomas, although the magnitude of their contribution to the disease process of HMO is uncertain.1-3 The median age of diagnosis is 3 years.4 Of note, early diagnosis can be difficult because small lesions are difficult to detect on plain films, age-dependent penetrance, and variable expressiveness. Mutations in EXT1 are often associated with more severe manifestations of the syndrome, but features associated with positive prognosis include female sex, involvement of less than 5 sites, and EXT2 mutation.1,3,4 Almost all cases are identified by age 12 years, with male predominance and an estimated prevalence of 1 in 50,000 population.1-5
Diagnostic criteria are met when there are 2 or more osteochondromas in the appendicular and axial bones with lesions exhibiting a preponderance for the proximal and distal femur and tibia, proximal humerus, distal radius, and distal ulna, as seen in our patient.1
EXT1 and EXT2 produce enzymes involved in posttranslational modifications required to produce functional heparan sulfate. Heparan sulfate is a ubiquitous glycosaminoglycan expressed on the cell surfaces of many tissues. In bone, heparan sulfate acts via numerous extracellular mechanisms to regulate growth factor gradients, such as fibroblast-growth factor, which are involved in bone growth and development.6,7 Disruption in this signaling system is thought to play a key role in the pathophysiology of the disorder.8 Disruption of the extracellular function of heparan sulfate has also shown profound effects in the central nervous system, with aberrancy in neural proliferation, migration, and differentiation noted in postmortem analysis of the subventricular zone of the lateral ventricles of individuals with autism-spectrum disorders.9,10
This association has also been displayed in rodent models, with subjects demonstrating disturbances of behavior commonly displayed by individuals with autism spectrum disorders, including difficulties engaging in communication and social behavior, as well as engaging in repetitive behaviors.11 Further investigation may yield insight into the mechanisms of our patient’s developmental delays.
Prognosis is varied, with osteochondromas typically ceasing growth upon maturity of the patient’s skeleton. The table lists possible complications from the development of osteochondromas cited in the literature.1,3,5,12-28 Due to the possibility of morbidity from various complications related to the condition, it is important that providers caring for patients with HMO relay to parents the importance of monitoring to facilitate early intervention.
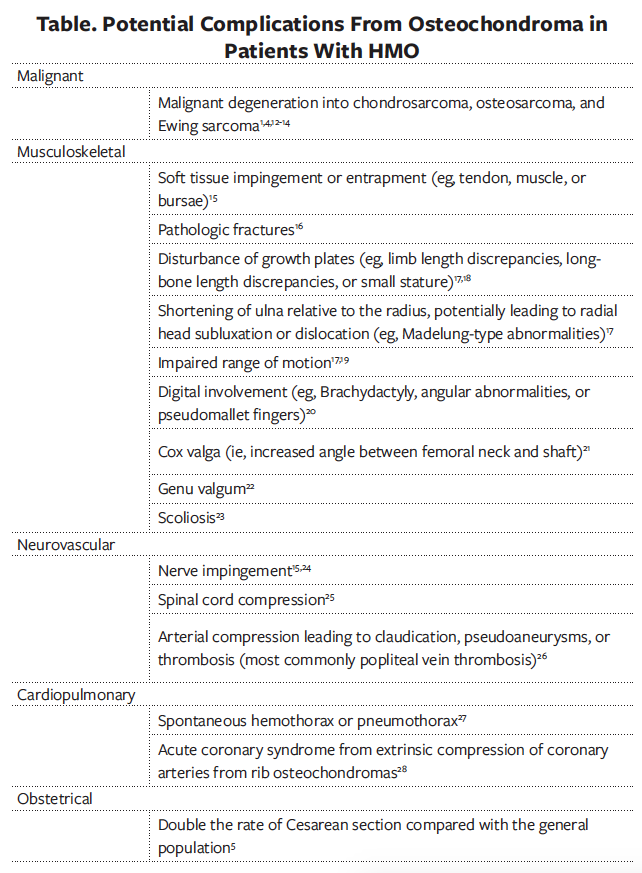
Depending on the size and location of the osteochondromas, the lesions can predispose the patient to pain, pathologic fractures, pneumothorax, compression of the spinal cord, and growth abnormalities from the involvement of the epiphyseal plates in younger, developing patients.15-20,25,27 Malignant transformation into chondrosarcoma, typically in young adulthood, has been estimated to be approximately 2.7% to 5.0%.3,12 Patients should be monitored for progression with screening imaging, followed by advanced imaging if warranted. Tumor characteristics associated with an increased risk of malignant transformation include continued growth after skeletal maturity and location, with an increased likelihood of transformation in lesions of the spine, scapula, pelvis, and proximal femur.12
Patient outcome. The patient’s parents were offered genetic testing to further elaborate on the causative mutation and its implications regarding prognosis, but they declined. The patient now has a younger sibling who does not show any signs or symptoms of HMO or other genetic syndromes. Lack of genetic testing leaves the patient’s disease process potentially attributable to other causes, such as metachondromatosis, Langer-Giedion syndrome, or fibrodysplasia ossificans progressive. However, given the characteristic appearance and distribution of lesions, HMO is, and continues to be, the working clinical diagnosis.
The patient, now aged 6 years, is scheduled for annual radiography scans. A pediatric radiologist involved with this case report suggested that the nature and location of the palpable abnormalities on the left lower lateral ribcage could likely go undetected with the single anteroposterior view of the thorax. Should the patient develop signs or symptoms potentially related to these palpable masses or if parental concern persists, then oblique views of the thorax could be considered.
This patient has exhibited global developmental delay, and autism spectrum disorder was diagnosed. Specific delays in achieving age-appropriate milestones have included persistent difficulty in forming and maintaining a functional grasp, difficulty in manipulating tools/utensils, delayed progression of vocabulary and percentage of speech understandable by others, difficulties with impulse control (eg, engaging in biting, spitting, and kicking behaviors when frustrated), and in sensory processing, as he is noted by his parents and providers to be unable to tolerate environments with loud sounds or high levels of activity. He has received occupational therapy and speech therapy and has made notable progress. He is progressively working toward achieving milestones typically achieved by neurotypical children aged 3 to 5 years.
1. Pannier S, Legeai-Mallet L. Hereditary multiple exostoses and enchondromatosis. Best Pract Res Clin Rheumatol. 2008;22(1):45-54. https://doi.org/10.1016/j.berh.2007.12.004
2. Wuyts W, Van Hul W, De Boulle K, et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am J Hum Genet. 1998;62(2):346-354. https://doi.org/10.1086/301726
3. Pedrini E, Jennes I, Tremosini M, et al. Genotype-phenotype correlation study in 529 patients with multiple hereditary exostoses: identification of “protective” and “risk” factors. J Bone Joint Surg Am. 2011;93(24):2294-2302. https://doi.org/10.2106/jbjs.j.00949
4. Schmale GA, Conrad 3rd EU, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg Am. 1994;76(7):986-992. https://doi.org/10.2106/00004623-199407000-00005
5. Wicklund CL, Pauli RM, Johnston D, Hecht JT. Natural history study of hereditary multiple exostoses. Am J Med Genet. 1995;55(1):43-46. https://doi.org/10.1002/ajmg.1320550113
6. Yan D, Lin X. Shaping morphogen gradients by proteoglycans. Cold Spring Harb Perspect Biol. 2009;1(3):a002493. https://doi.org/10.1101/cshperspect.a002493
7. Kelleher FC, O’Sullivan H, Smyth E, McDermott R, Viterbo A. Fibroblast growth factor receptors, developmental corruption and malignant disease. Carcinogenesis. 2013;34(10):2198-2205. https://doi.org/10.1093/carcin/bgt254
8. Beltrami G, Ristori G, Scoccianti G, Tamburini A, Capanna R. Hereditary multiple exostoses: a review of clinical appearance and metabolic pattern. Clin Cases Miner Bone Metab. 2016;13(2):110-118. https://doi.org/10.11138/ccmbm/2016.13.2.110
9. Pearson BL, Corley MJ, Vasconcellos A, Blanchard DC, Blanchard RJ. Heparan sulfate deficiency in autistic postmortem brain tissue from the subventricular zone of the lateral ventricles. Behav Brain Res. 2013;243:138-145. https://doi.org/10.1016/j.bbr.2012.12.062
10. Li H, Yamagata T, Mori M, Momoi MY. Association of autism in two patients with hereditary multiple exostoses caused by novel deletion mutations of EXT1. J Hum Genet. 2002;47(5):262-265. https://doi.org/10.1007/s100380200036
11. Irie F, Badie-Mahdavi H, Yamaguchi Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc Natl Acad Sci USA. 2012;109(13):5052-5056. https://doi.org/10.1073/pnas.1117881109
12. Czajka CM, DiCaprio MR. What is the proportion of patients with multiple hereditary exostoses who undergo malignant degeneration? Clin Orthop Relat Res. 2015;473(7):2355-2361. https://doi.org/10.1007/s11999-015-4134-z
13. Angelini A, Guerra G, Mavrogenis AF, Pala E, Picci P, Ruggieri P. Clinical outcome of central conventional chondrosarcoma. J Surg Oncol. 2012;106(8):929-937. https://doi.org/10.1002/jso.23173
14. Marrero Barrera PA, Marrero Ortiz PV. Ewing sarcoma superimposed on a previous osteochondroma in multiple osteochondromatosis. Orthopedics. 2014;37(4):e403-e406. https://doi.org/10.3928/01477447-20140401-65
15. Darilek S, Wicklund C, Novy D, et al. Hereditary multiple exostosis and pain. J Pediatr Orthop. 2005;25(3):369-376. https://doi.org/10.1097/01.bpo.0000150813.18673.ad
16. Robbins MM, Kuo S, Epstein R. Non-traumatic fracture of an osteochondroma mimicking malignant degeneration in an adult with hereditary multiple exostoses. Radiol Case Rep. 2015;3(3):99. https://doi.org/10.2484/rcr.v3i3.99
17. Ali S, Kaplan S, Kaufman T, Fenerty S, Kozin S, Zlotolow DA. Madelung deformity and Madelung-type deformities: a review of the clinical and radiological characteristics. Pediatr Radiol. 2015;45(12):1856-1863. https://doi.org/10.1007/s00247-015-3390-0
18. Staal HM, Goud AL, van der Woude HJ, et al. Skeletal maturity of children with multiple osteochondromas: Is diminished stature due to a systemic influence? J Pediatr Orthop. 2015;9(5):397-402. https://doi.org/10.1007/s11832-015-0680-x
19. Noonan KJ, Feinberg JR, Levenda A, Snead J, Wurtz LD. Natural history of multiple hereditary osteochondromatosis of the lower extremity and ankle. J Pediatr Orthop. 2002;22(1):120-124. https://journals.lww.com/pedorthopaedics/Abstract/2002/01000/Natural_History_of_Multiple_Hereditary.25.aspx
20. Woodside JC, Ganey T, Gaston RG. Multiple osteochondroma of the hand: initial and long-term follow-up study. Hand (N Y). 2015;10(4):616-620. https://doi.org/10.1007/s11552-015-9775-6
21. Wang YZ, Park KW, Oh CS, et al. Developmental pattern of the hip in patients with hereditary multiple exostoses. BMC Musculoskelet Disord. 2015;16:54. https://doi.org/10.1186/s12891-015-0514-5
22. Clement ND, Porter DE. Can deformity of the knee and longitudinal growth of the leg be predicted in patients with hereditary multiple exostoses? A cross-sectional study. Knee. 2014;21(1):299-303. https://doi.org/10.1016/j.knee.2012.10.029
23. Matsumoto Y, Matsumoto K, Harimaya K, Okada S, Doi T, Iwamoto Y. Scoliosis in patients with multiple hereditary exostoses. Eur Spine J. 2015;24(7):1568-1573. https://doi.org/10.1007/s00586-015-3883-4
24. Demiroğlu M, Özkan K, Kılıç B, Akçal A, Akkaya M, Özkan FÜ. Deep peroneal nerve palsy due to osteochondroma arising from fibular head and proximal lateral tibia. Int J Surg Case Rep. 2017;31:200-202. https://doi.org/10.1016/j.ijscr.2017.01.050
25. Sciubba DM, Macki M, Bydon M, et al. Long-term outcomes in primary spinal osteochondroma: a multicenter study of 27 patients. J Neurosurg Spine. 2015;22(6):582-588. https://doi.org/10.3171/2014.10.spine14501
26. Nasr B, Albert B, David CH, da Fonseca PM, Badra A, Gouny P. Exostoses and vascular complications in the lower limbs: two case reports and review of the literature. Ann Vasc Surg. 2015;29(6):1315.e1317-1315.e1314. https://doi.org/10.1016/j.avsg.2015.02.020
27. Ravindran R, Jordan S, Bush A. An extra piece of grey. Thorax. 2015;70(7):705-706. https://doi.org/10.1136/thoraxjnl-2015-207061
28. Rodrigues JCL, Mathias HC, Lyen SM, et al. A novel cause of acute coronary syndrome due to dynamic extrinsic coronary artery compression by a rib exostosis: multimodality imaging diagnosis. Can J Cardiol. 2015;31(10):1303.e9-1303.e11. https://doi.org/10.1016/j.cjca.2015.05.008