
Peer Reviewed
Creutzfeldt-Jakob Disease: A Review of Recognition and Management
Authors:
Alison Patrick, MPAS, PA-C, and Pamela Wireman, MPAS, PA-C
Kettering College, Kettering, Ohio
Citation:
Patrick A, Wireman P. Creutzfeldt-Jakob disease: a review of recognition and management [published online September 18, 2018]. Neurology Consultant.
ABSTRACT: Although Creutzfeldt-Jakob disease (CJD) is a rare disease, its presentation often is similar to that of many other neurologic diseases, resulting in a high rate of misdiagnosis. CJD is characterized by a rapidly progressive dementia along with other neurologic symptoms. Diagnosis is based on clinical findings, magnetic resonance imaging, and laboratory test results. Although no treatment exists, affected patients should be provided with appropriate supportive care.
KEYWORDS: Creutzfeldt-Jakob disease, prions, transmissible spongiform encephalopathy, dementia
Creutzfeldt-Jakob disease (CJD) belongs to a rare class of diseases known as prion diseases or transmissible spongiform encephalopathies (TSEs). Also included in this class are Gerstmann-Sträussler-Scheinker disease and fatal familial insomnia in humans and bovine spongiform encephalopathy (BSE; commonly known as mad cow disease) and chronic wasting disease in animals.1 CJD is the most common prion disease among humans, with an approximate worldwide incidence of 1 in 1 million people per year and affecting approximately 300 people in the United States each year.2,3 Prion diseases are characterized by their long incubation periods and rapid progression to death once symptoms appear. Symptom onset usually begins between the ages of 50 and 70 years in most patients, with death following within 1 year.2,4,5
Although CJD is uncommon, the ability to quickly identify features of the disease is paramount. Many cases of CJD are incorrectly diagnosed initially due to symptoms that mimic more common diseases (eg, viral encephalitis, Alzheimer disease, paraneoplastic disorders) and primary care providers’ and neurologists’ failure to include prion diseases in the differential diagnosis.6 In one retrospective study, only 18% of patients with CJD had received a correct diagnosis upon initial assessment.6 The mean time from onset of symptoms to diagnosis was approximately 8 months; thus, patients had been two-thirds through the disease course before a correct diagnosis had been made.6 Moreover, two-thirds of radiology reports failed to recognize the pathognomonic feature of CJD on magnetic resonance imaging (MRI) scans.6 Patients with CJD often undergo a barrage of tests and receive any of a multitude of misdiagnoses at a time when they and their family members should be focusing on quality of life, end-of-life planning, and potential enrollment in clinical trials.
Pathophysiology
Although its cause is not completely understood, CJD is thought to result from a prion protein (PrP) changing to an abnormal, potentially transmissible form (PrPSc).7,8 The transmissible form is atypically folded and induces normal prion proteins to misfold through direct contact.7,8 These misfolded proteins can then induce surrounding prion proteins to misfold, thereby causing a chain reaction.7,8 The misfolded proteins aggregate extracellularly within the central nervous system, forming structures called amyloid plaques.7,8 These plaques cause sponge-like holes to form in neural tissue, which alter normal tissue structure and function.7,8 The result is neuronal death and synaptic loss.7,8 Over the past 10 years, researchers have shown that this mechanism (ie, normal proteins converted to an abnormal form) might also be responsible for more common neurodegenerative diseases such as Parkinson disease and Alzheimer disease.2
Risk Factors
Most cases of CJD occur sporadically (sCJD), meaning that the trigger prion protein spontaneously misfolds in the affected individual, and no prior transmission pattern is identified. Although no risk factors have been identified for this form of CJD, sCJD occurs worldwide and accounts for 85% of CJD cases. For this reason, sCJD is the focus of this article.
Nevertheless, CJD can be acquired through means other than spontaneous mutation. When the misfolded prion protein results from a mutated prion gene, the gene can be passed down to family members (familial CJD). Furthermore, the disease can be transferred iatrogenically from an affected person to an unaffected person by way of medical procedures (typically neurosurgical procedures). Variant CJD (vCJD) occurs when an abnormal prion protein is transferred from cattle to humans.4
Precautions
Regardless of how they are acquired, all types of CJD (including sCJD) are transmissible.9 The disease can be transmitted to other patients if the mutated prion protein is transferred from the affected individual's brain to an unaffected individual's brain via contaminated surgical equipment. Therefore, appropriate precautions should be taken in individuals with the disease, such as avoiding brain procedures and/or following appropriate decontamination procedures in the operating room, including autoclaving instruments at higher temperatures than would normally be used for sterilization, immersing contaminated instruments in sodium hydroxide, and discarding used instruments.10
Because of the risk of probable bloodborne transmission of vCJD, the American Red Cross has adopted policies that include deferral of potential blood donors who have lived in or visited areas where BSE rates are high.11 This includes anyone living in or visiting the United Kingdom for more than 3 months since 1980, or living in or visiting Oman, Turkey, or another European country (besides the United Kingdom) for more than 6 months since 1980.11
Due to the risk of bovine–human transmission of CJD, the US Department of Agriculture has taken steps to safeguard food. These safeguards include banning the slaughter of “downer cattle” (ie, cattle that should rise on examination but do not) for consumption, holding the carcasses of animals tested for BSE until the results are confirmed negative, and banning the removal of the last traces of skeletal muscle from certain areas of the brain, eyes, spinal cord, and vertebral column from cattle 30 months of age or older.12 This also includes banning the recovery of meat from the tonsils and small intestines of all cattle.12
Clinical Manifestations
CJD affects many areas of the brain, so its presentation can mimic other neurologic and psychiatric disorders, including Alzheimer disease, Parkinson disease, and Huntington disease. However, unlike these conditions, sCJD is rapidly progressive once symptoms develop.2 Patients often report vague symptoms at first, including malaise, anxiety, mood changes, dizziness, and difficulty concentrating. These initial symptoms are then promptly followed by more obvious cognitive symptoms including amnesia, confusion, and impaired attention. These symptoms also can be accompanied by visual disturbances, aphasia, apraxia, and spontaneous or induced myoclonus.2,13 Approximately one-third of patients present with ataxia in addition to cognitive impairment.
Diagnosis
Although brain biopsy is the gold standard for the diagnosis of sCJD, it is often unnecessary and can potentially be dangerous due to the possibility of transmitting the abnormal prion proteins from contaminated surgical equipment.4 Instead, the Centers for Disease Control and Prevention (CDC) recommends that a “probable” diagnosis be made on the basis of certain clinical features, electroencephalography (EEG) findings, cerebrospinal fluid (CSF) analysis results, and MRI findings.14
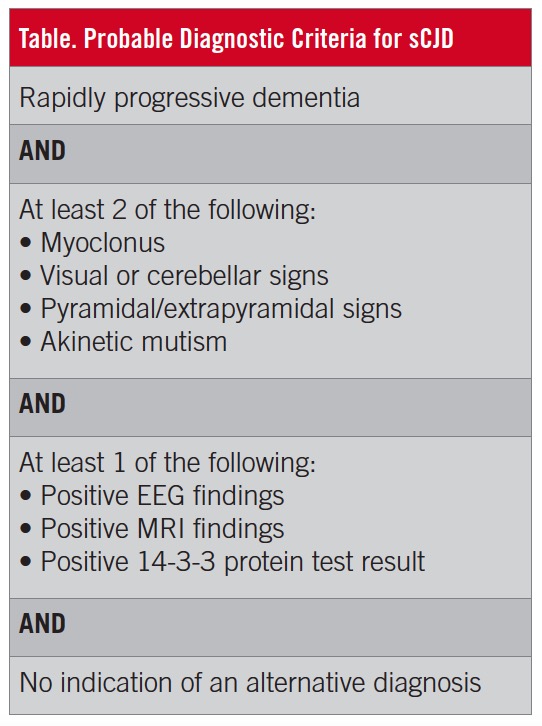
The CDC recommends that the following criteria be used for diagnosis: First, patients must present with rapidly progressive dementia and 2 of the following features: myoclonus, visual or cerebellar signs, pyramidal or extrapyramidal signs, and akinetic mutism (Table). Along with these clinical findings, patients also must have at least 1 of the following results after testing: a finding of periodic sharp wave complexes on EEG, a positive 14-3-3 protein test result on CSF analysis, or abnormal signals in the putamen and/or head of the caudate nucleus on MRI using diffusion-weighted imaging or fluid-attenuated inversion recovery.4,14 Along with these findings, there must be no indication of an alternative diagnosis.14
It is important to note, however, that none of the above tests are specific for sCJD, and many patients go undiagnosed despite these tests having been performed. Because of this, a number of laboratories have established a newer approach to diagnosing sCJD known as real-time quaking-induced conversion (RT-QuIC).5 This method involves obtaining a sample of CSF or cells deep within the patient’s nostril and mixing it with normal recombinant prion proteins (rPrP). The test results are positive for sCJD when the sample induces the rPrP substrate to aggregate.5 Unlike the diagnostic criteria recommended by the CDC and the World Health Organization, which involve several tests and provide a specificity of only 70.8%, this technique involves only 1 laboratory test and is highly sensitive (85.7%) and highly specific (100%).5,15 Although RT-QuIC is not yet available in many laboratories, the test is routinely performed at prion disease surveillance centers in several countries.4 Modified versions of the test also are being developed to improve identification of Parkinson disease and Alzheimer disease, which are currently diagnosed based on clinical findings alone.16
Management
No effective treatment exists for CJD. The disease is uniformly fatal, with most patients declining into a vegetative state followed by death within a year after the onset of initial symptoms.2,4,5 Management of CJD is therefore supportive.3 Opioid medications can help relieve any associated pain, and drugs such as sodium valproate and clonazepam may be used to help relieve myoclonus.3 Feeding and ventilator support may be required in some patients.17 Because CJD is rapidly progressive once symptoms begin, end-of-life care including advanced directives, living arrangements, and referral to palliative care should be discussed soon after diagnosis.17 Enrollment in research and clinical trials can be considered.
Several immunologic approaches are emerging as potential therapeutic options for CJD. For instance, anti-prion antibody fragments that bind to misfolded prion proteins may help prevent their propagation. Studies are showing promise in cell and animal trials, but they have yet to be tested in humans.18
Take-Home Message
On average, sCJD is misdiagnosed 4 times before a patient receives the correct diagnosis.6 Early diagnosis of sCJD is essential not only for the patient and family members in order to provide quality end-of-life care, but also to prevent possible iatrogenic transmission of the disease through blood or organ donation, surgical equipment, or medical procedures.6 Therefore, sCJD should be included in the differential diagnosis for any patient presenting with rapidly progressive dementia, and appropriate workup should be undertaken. Furthermore, CJD should be considered in any patient presenting with symptoms of Alzheimer disease, Huntington disease, or Parkinson disease.6
- Prion diseases. Centers for Disease Control and Prevention. https://www.cdc.gov/prions/index.html. Updated August 17, 2017. Accessed September 18, 2018.
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618-628.
- Creutzfeldt-Jakob disease fact sheet. National institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Creutzfeldt-Jakob-Disease-Fact-Sheet. Updated August 21, 2018. Accessed September 18, 2018.
- Brown HG, Lee JM. Creutzfeldt-Jakob disease. UpToDate. https://www.uptodate.com/contents/creutzfeldt-jakob-disease. Updated June 16, 2017. Accessed September 18, 2018.
- McGuire LI, Poleggi A, Poggiolini I, et al. Cerebrospinal fluid real-time quaking-induced conversion is a robust and reliable test for sporadic Creutzfeldt-Jakob disease: an international study. Ann Neurol. 2016;80(1):160-16
- Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol. 2012;69(12):1578-1582.
- Frosch MP, Anthony DC, De Girolami U. Prion diseases. In: Kumar V, Abbas AK, Aster JC, eds. Robbins and Cotran Pathologic Basis of Disease. 9th ed. Philadelphia, PA: Elsevier Saunders; 2015:1281-1282.
- du Plessis DG. Prion protein disease and neuropathology of prion disease. Neuroimaging Clin North Am. 2008;18(1):163-182.
- Brown P, Gibbs CJ Jr, Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35(5):513-52
- Rutala WA, Weber DJ; Society for Healthcare Epidemiology of America. Guideline for disinfection and sterilization of prion-contaminated medical instruments. Infect Control Hosp Epidemiol. 2010;31(2):107-117.
- Kleinman S. Blood donor screening: medical history. UptoDate. https://www.uptodate.com/contents/blood-donor-screening-medical-history. Updated March 21, 2018. Accessed September 18, 2018.
- Brown HG, Lee JM. Variant Creutzfeldt-Jakob disease. UptoDate. https://www.uptodate.com/contents/variant-creutzfeldt-jakob-disease. Updated June 16, 2017. Accessed September 18, 2018.
- Krasnianski A, Bohling GT, Heinemann U, et al. Neuropsychological symptoms in sporadic Creutzfeldt-Jakob disease patients in Germany. J Alzheimers Dis. 2017;59(1):329-337.
- CDC's diagnostic criteria for Creutzfeldt-Jakob disease (CJD), 2010. Centers for Disease Control and Prevention. https://www.cdc.gov/prions/cjd/diagnostic-criteria.html. Updated February 11, 2015. Accessed September 18, 2018.
- Newey CR, Sarwal A, Wisco D, Alam S, Lederman RJ. Variability in diagnosing Creutzfeldt‐Jakob disease using standard and proposed diagnostic criteria. J Neuroimaging 2013;23(1):58-63.
- Fairfoul G, McGuire LI, Pal S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3(10):812-818.
- Creutzfeldt-Jakob Disease. National Health Service. https://www.nhs.uk/conditions/creutzfeldt-jakob-disease-cjd/treatment. Updated June 14, 2018. Accessed September 18, 2018.
- Cardinale A, Biocca S. Gene-based antibody strategies for prion diseases. Int J Cell Biol. 2013;2013:710406. doi:10.1155/2013/710406