
Peer Reviewed
Alpers-Huttenlocher Syndrome
AUTHORS:
Nitish Chourasia, MD1 • Dillon Burks, MD2 • Meenakshi Bhattacharjee, MD3 • Rajan Patel, MD4 • Anand Gourishankar, MBBS, MRCP, MAS2
AFFILIATIONS:
1Division of Child and Adolescent Neurology, McGovern Medical School at UTHealth, Houston, Texas
2Department of Pediatrics, McGovern Medical School at UTHealth, Houston, Texas
3Department of Neuropathology, McGovern Medical School at UTHealth, Houston, Texas
4Department of Radiology, McGovern Medical School at UTHealth, Houston, Texas
CITATION:
Chourasia N, Burks D, Bhattacharjee M, Patel R, Gourishankar A. Alpers-Huttenlocher syndrome. Consultant. 2020;60(5):e9. doi:10.25270/con.2020.03.00018
Received January 15, 2020. Accepted February 27, 2020.
DISCLOSURES:
The authors report no relevant financial relationships.
CORRESPONDENCE:
Anand Gourishankar, MBBS, MRCP, MAS, Division of Pediatric Hospital Medicine, McGovern Medical School at UTHealth, 6431 Fannin St Ste 732, Houston TX 77030 (anand.gourishankar@uth.tmc.edu)
A 9-month-old boy with a history of developmental delay, failure to thrive, and intractable epilepsy of unknown etiology presented to the hospital with twitching of his right arm and face, concerning for seizures. He received a dose of lorazepam followed by loading doses of fosphenytoin and phenobarbital, which did not alleviate the seizures. He was subsequently intubated and transferred to the pediatric intensive care unit for further management of his seizures.
The boy had been born to nonconsanguineous parents. He had no significant family history of neurological disorders, and he had a normal 3-year-old sister.
His vital signs at admission were as follows: temperature, 36.2°C; pulse rate, 138 beats/min; blood pressure, 90/45 mm Hg; respiratory rate, 32 breaths/min; and oxygen saturation, 98% with endotracheal intubation. His weight was 5.6 kg, his length was 60 cm, and his head circumference was 41 cm; all 3 of these values were below the third percentile. On examination, he was normocephalic, and he had equal and reactive pupils. Breath sounds were symmetric bilaterally. His heart rate was regular, with no discernible murmurs. He had a nasogastric tube in place, and the abdomen was soft, nontender, and nondistended. Neurologic examination showed intact cranial nerve function, decreased tone throughout, normal muscle bulk, and symmetric reflexes. He was not making any audible sounds for his age and was not yet rolling over.
The patient had had an initial seizure onset at 5 months of age, and before the current hospitalization, he had previously been admitted twice to the hospital for worsening seizures. Results of extensive laboratory evaluations for epilepsy—including chromosomal microarray, cerebrospinal fluid (CSF) neurotransmitters, pyruvate, lactic acid, and metabolic panels including urine organic acids, acylcarnitine profile, and plasma amino acids—had been unremarkable. Brain magnetic resonance imaging (MRI) abnormalities involving distinct regions—deep posterior subcortical white matter and frontal, insular, temporal areas—were noted in each of the previous 2 admissions, respectively (Figure 1). The etiology of the epilepsy and developmental delay remained unclear. There was no concern for infection.
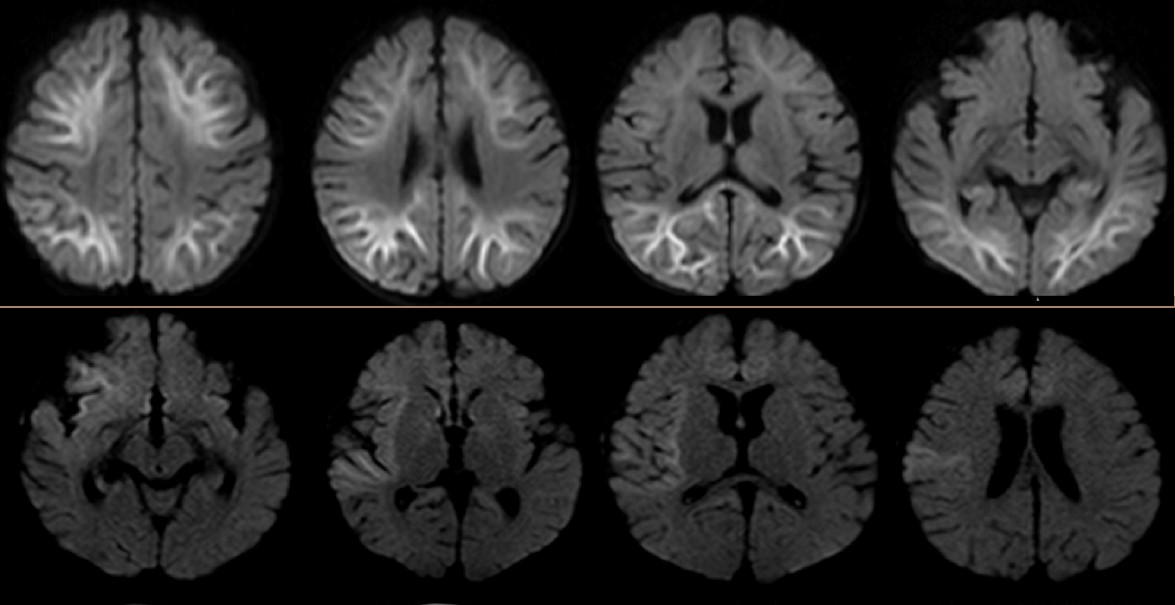
Figure 1. Top: Abnormal restricted diffusion involving both cerebral hemispheres with predominant involvement of deep and subcortical white matter. Bottom: Abnormal restricted diffusion involving the right insular region, right frontal region, right temporal operculum, and right amygdala of the brain.
An electroencephalogram (EEG) during the current admission showed frequent multifocal epileptiform discharges, which was a finding similar to that of his prior EEGs. Since he had continued to have breakthrough seizures despite 3 antiepileptic drugs (phenobarbital, levetiracetam, and lacosamide), a fourth antiepileptic drug (clobazam) was added. The seizures improved, but his liver function test (LFT) results, which included alanine aminotransferase (ALT), aspartate aminotransferase (AST), and γ-glutamyltransferase (GGT), and his lipase level, showed abnormal elevations for 3 days after addition of clobazam (Figure 2).
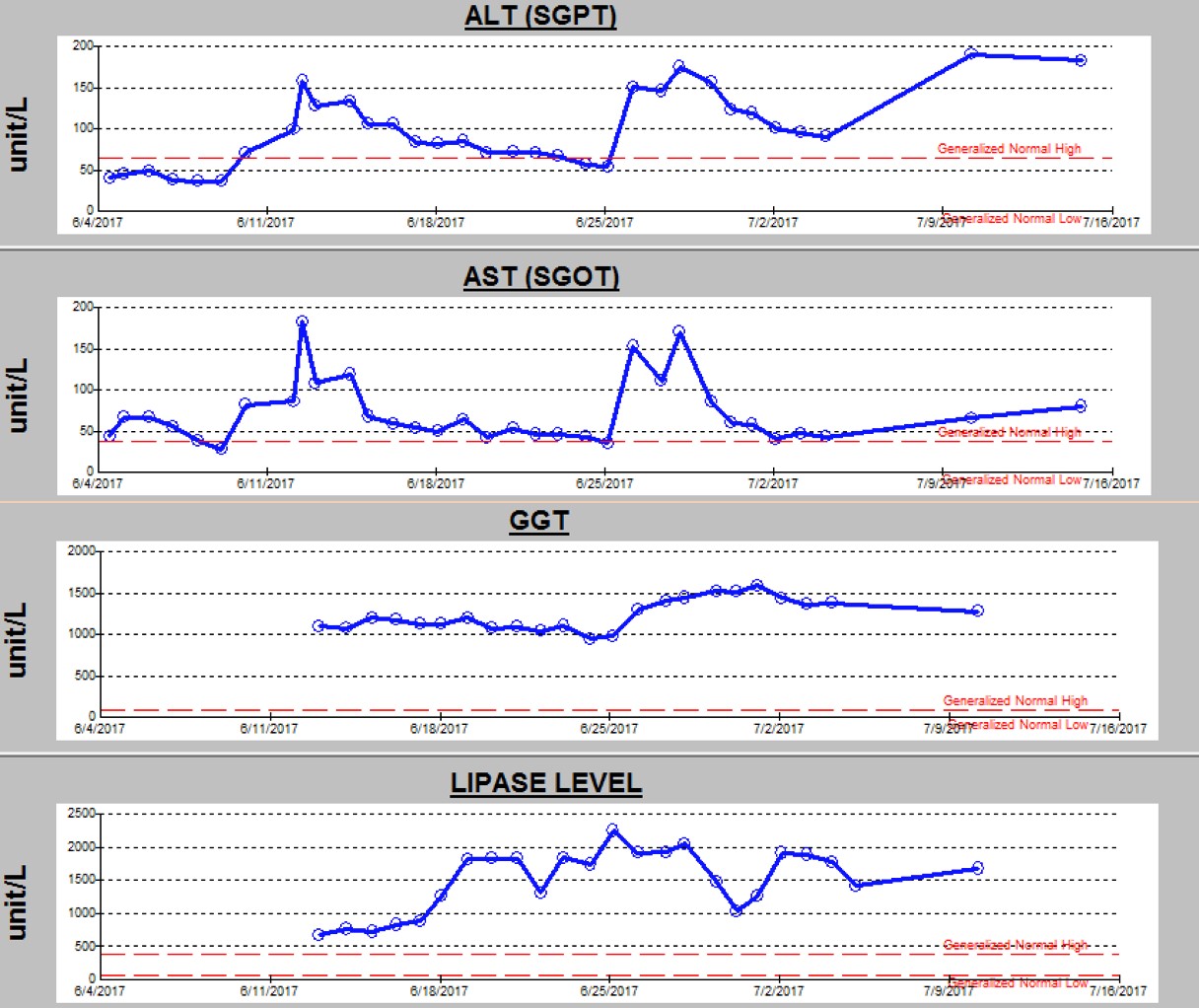
Figure 2. Trend of ALT, AST, GGT, and lipase levels during the current hospital stay.
Clobazam was discontinued out of concern for possible drug-related adverse effects, but LFT results continued to remain elevated. The serum amylase level and abdominal ultrasonography findings were normal, which ruled out pancreatitis and other abnormalities of the hepatobiliary system. MRI with magnetic resonance spectroscopy of the brain (Figure 3) demonstrated a new area of involvement in the left occipital lobe.
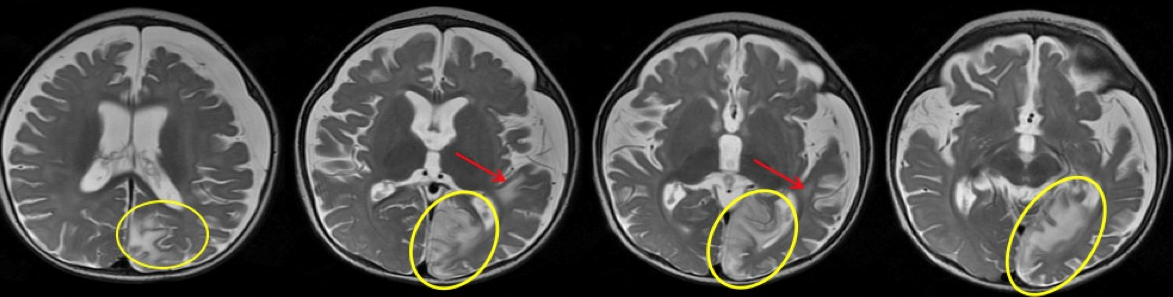
Figure 3. New T2 hyperintensity involving the left occipital lobe and left angular/supramarginal region without associated restricted diffusion. Interval increased cerebral volume loss and development of bilateral subdural fluid collections are present, left greater than right.
Muscle biopsy was performed, the results of which showed mildly increased and enlarged mitochondria on electron microscopy (Figure 4).
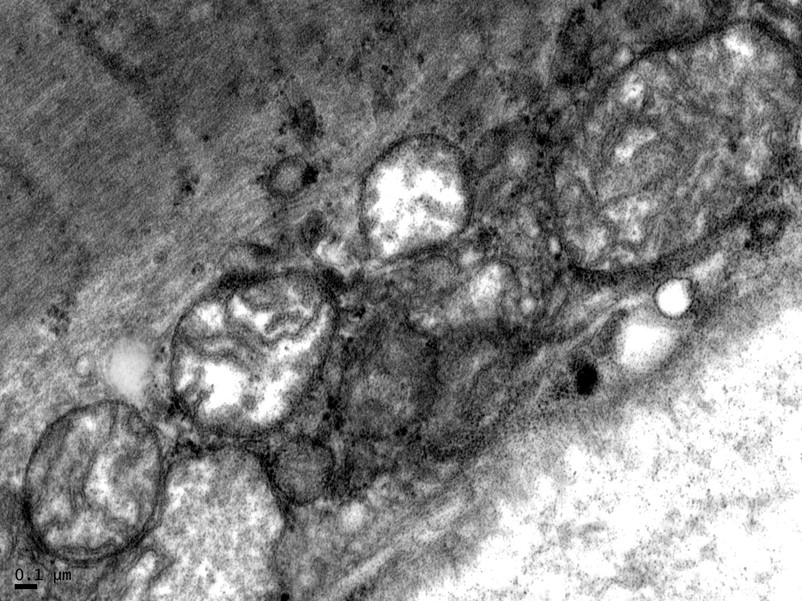
Figure 4. Muscle biopsy results showing enlarged mitochondria on electron microscopy.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of intractable epilepsy, developmental delay, and failure to thrive in infancy includes a broad range of metabolic and genetic conditions, including mitochondrial disorders, glycine encephalopathy, urea cycle disorders, cholesterol metabolism disorders, peroxisomal disorders, glucose transporter type 1 deficiency syndrome, lysosomal disorders, carbohydrate metabolism disorders, and congenital disorders of glycosylation, among others.
Liver dysfunction in infants in the setting of intractable epilepsy and developmental delay narrows the diagnosis to urea cycle disorders (arginase deficiency), peroxisomal disorders (Zellweger syndrome), amino acid metabolism (tyrosinemia), carbohydrate metabolism (galactosemia, fructosemia) and mitochondrial disorders (Alpers-Huttenlocher syndrome, or AHS).
Based on the clinical presentation and the progression of symptoms, including liver dysfunction and abnormal brain MRI findings, our patient received a diagnosis of AHS. Mitochondrial disease process was first suspected when multiple regions of the brain not conforming to vascular territories were noted; the diagnosis of AHS was confirmed once liver dysfunction occurred. Our patient fits the clinical diagnostic criteria, which include the presence of the classical triad (liver dysfunction, intractable seizures, and developmental delay/regression) along with 2 of 11 additional clinical findings (Table).

Although AHS is a rare disorder, there is an overlap of Alpers-like disorders without liver disease with mutation of the DNA polymerase γ gene (POLG), and less than a third of persons with AHS may not have a POLG mutation. In our patient’s case, the neurologist deferred genetic testing given the clinical certainty of the diagnosis and the parents’ preference for no further testing.
DISCUSSION
AHS is a heterogeneous group of diseases that feature progressive cerebral deterioration and liver disease.1 The combination of seizures at a young age, developmental delay or regression, and worsening liver function are typical symptoms of AHS, an autosomal recessive neurodegenerative pathological process originating in the mitochondria.2 Bernard J. Alpers3 first described the condition in 1931, and later Peter Huttenlocher4 expanded on the phenotypes.
Within the mitochondria, nuclear and mitochondrial DNA products and proteins combine to form complexes I through IV of the electron transport chain (ETC). AHS classically occurs when mutations in the POLG render mitochondrial polymerase-γ ineffective. This gene provides instructions for making the active portion (the α subunit) of the protein polymerase γ. If the function of polymerase γ becomes compromised, mitochondrial DNA products can begin to malfunction due to incorrectly incorporated base pairs and mismatches. This compromise, in turn, can alter the efficiency and capability of the ETC complexes using mitochondrial DNA.5
The organ systems most at risk of complications in AHS are those with the most substantial need of energy: the liver, the central nervous system, and the peripheral nervous system. ETC and enzyme evaluation may provide clues to a diagnosis early in the disease, but variability (from normal chains to multiple defects) in results excludes this screening as a diagnostic tool. Currently, researchers are studying the natural history of AHS, following patients’ cases to measure the age of onset of symptoms vs POLG mutation type.6
AHS has a bimodal distribution, with cases reported in patients between the ages of 3 months and 8 years and again between the ages of 17 and 24 years, in which it additionally presents with dementia.2 Eighty percent of patients develop symptoms in the first 2 years of life. The occurrence rate of AHS is 1 in 100,000.1 Newborns are asymptomatic and show normal development until a few weeks or years of age. The difference between these 2 populations is thought to lie in whether or not the patient possesses a heterozygous recessive mutation (which presents in childhood) or a homozygous recessive mutation (which presents in later years). The speed of neurodegeneration typically hastens after the onset of seizures, with death commonly occurring within 4 years of onset. As the disease progresses, these seizures may become resistant to traditional epilepsy treatments. Other neurological findings include headaches (migraine with visual symptoms), blindness, encephalopathy, neuropathies, choreoathetosis, myoclonus, and ataxia.1,2
Cardiomyopathy also may occur.2 Infants may present with failure to thrive (as in our patient’s case) and repeated infections.2 Liver involvement is one of the defining features of AHS. The process of liver failure may worsen as a result of the disease itself, and it may worsen with the antiepileptic drug valproic acid, a catalyst for liver dysfunction in these patients.2 Hypoglycemia, coagulopathies, cirrhosis, bridging fibrosis, and bile duct involvement are a few of the hepatic manifestations observed in patients with AHS.1,2,4
MANAGEMENT
Treatment of AHS is limited to symptom management and supportive care. Palliation should be a central focus of care once a diagnosis has been made. The disease inevitably leads to fatal hepatic failure and encephalopathy. Neurological manifestations such as seizures should be treated with anti-epileptics, although seizures become refractory to antiepileptic medications in the later stages of the disease. Valproic acid is contraindicated. Anecdotally, a ketogenic diet may control seizures.7 Hypoglycemia secondary to liver dysfunction may be treated by continuous feeding via a gastric feeding tube or an increased frequency of meals; exercise caution, since gastrointestinal tract dysmotility is possible. Due to the progressive and irreversible neurological damage incurred throughout the disease, patients with AHS are not candidates for liver transplant.8
AHS has an abysmal prognosis. Death usually occurs within the first decade of life, and intractable seizures (leading to cardiorespiratory failure) may lead to death.
OUTCOME OF THE CASE
The diagnosis of AHS and its prognosis were discussed with the family in a multidisciplinary meeting.9 The patient was discharged home on hospice with anti-epileptic treatment to assist with seizure control, although excessive sedation is a complication, and high doses of an anti-epileptic do not provide complete seizure control. The boy remained on continuous enteral feeding, with a daily laxative. We recommended continuing physical therapy at home. He did not require noninvasive ventilation upon discharge. He went home on levocarnitine, which may help in the setting of liver failure and has low toxicity.
LESSONS FOR THE CLINICIAN
AHS should remain a suspicion for any child presenting with intractable seizures of unknown etiology. An MRI that shows multiple areas of evolving brain injury in subsequent scans should raise suspicion for mitochondrial encephalopathies. And valproic acid should not be initiated in any children under the age of 2 years or with seizures of unknown etiology until POLG evaluation has been completed to rule out a possible underlying mitochondrial abnormality.
REFERENCES:
- Rose HR, Al Khalili Y. Alpers-Huttenlochen syndrome (AHS, Alper disease). StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK540966/. Updated December 26, 2019. Accessed March 10, 2020.
- Saneto RP, Cohen BH, Copeland WC, Naviaux RK. Alpers-Huttenlocher syndrome. Pediatr Neurol. 2013;48(3):167-178. doi:10.1016/j.pediatrneurol.2012.09.014
- Alpers BJ. Diffuse progressive degeneration of the gray matter of the cerebrum. Arch Neurol Psychiatry. 1931;25(3):469-505. doi:10.1001/archneurpsyc.1931.02230030027002
- Huttenlocher PR, Solitare GB, Adams G. Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol. 1976;33(3):186-192. doi:10.1001/archneur.1976.00500030042009
- Chan SS, Copeland WC. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta. 2009;1787(5):312-319. doi:10.1016/j.bbabio.2008.10.007
- Alpers Huttenlocher Natural History Study (Alpers). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03034512. NCT03034512. Updated August 5, 2019. Accessed March 10, 2020.
- Joshi CN, Greenberg CR, Mhanni AA, Salman MS. Ketogenic diet in Alpers-Huttenlocher syndrome. Pediatr Neurol. 2009;40(4):314-316. doi:10.1016/j.pediatrneurol.2008.10.023
- Park S, Kang H-C, Lee J-S, Park YN, Kim S, Koh H. Alpers-Huttenlocher syndrome first presented with hepatic failure: can liver transplantation be considered as treatment option? Pediatr Gastroenterol Hepatol Nutr. 2017;20(4):259-262. doi:10.5223/pghn.2017.20.4.259
- Saneto RP. Alpers-Huttenlocher syndrome: the role of a multidisciplinary health care team. J Multidiscip Healthc. 2016;9:323-333. doi:10.2147/JMDH.S84900