
Genetics in Gastroenterology: What You Need to Know, Part 2
ABSTRACT: Many gastrointestinal diseases are inherited or have a genetic predisposition involved in disease expression. Recent research has uncovered the genes responsible for many of these conditions. Some conditions now have genetic testing available for diagnosis and for identifying asymptomatic family members. Certain genes have been associated with other diseases, but the development of the condition is not fully understood. These developments will continue to change how we diagnose and treat gastrointestinal conditions.
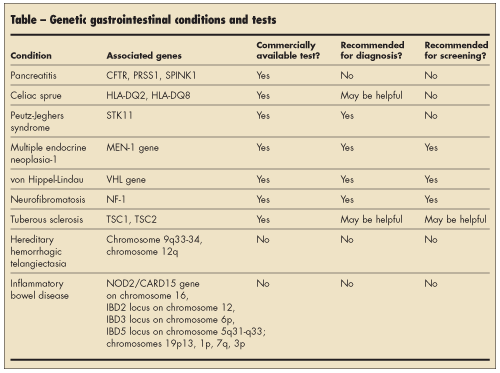
Many diseases involving the gastrointestinal organs are inherited or have a genetic predisposition involved in disease expression. Some of these have commercially available tests that can be used for diagnostic or screening purposes in predisposed individuals. Many others have a genetic component that has been identified.
These discoveries are already changing how we diagnose certain conditions and how we screen family members of patients with these conditions. In the future, we may be able to use this information to individually tailor therapies and improve outcomes and quality of life. The purpose of this 2-part article is to review this information and to discuss the implications for your practice. Here, in part 2, we focus on pancreatitis, celiac sprue, Peutz-Jeghers syndrome, neuroendocrine tumors, hereditary hemorrhagic telangiectasia, and inflammatory bowel disease. In a previous issue (CONSULTANT, February 2012, page 99), we addressed inherited colon cancer syndromes, juvenile polyposis, hereditary hemochromatosis, polycystic liver disease, autoimmune hepatitis, Budd-Chiari syndrome, alpha-1-antitrypsin deficiency, and Wilson disease.
PANCREATITIS
There is growing evidence that a significant number of patients with idiopathic pancreatitis, both acute and chronic, have an underlying genetic predisposition.1 The cystic fibrosis transmembrane conductance regulator (CFTR) gene is responsible for cystic fibrosis. It encodes for a cyclic adenosine monophosphate-dependent chloride channel that is located in the apical membrane of epithelial cells of the pancreas, intestine, liver, airway, vas deferens, and sweat glands. Complications arise from an inability to hydrate macromolecules within ductal lumens. In the pancreas, CFTR abnormalities cause obstruction of intralobular ducts by inspissated protein plugs. They may also have diminished bicarbonate secretion and therefore reduced alkalinization of the acinar lumen.
Before the CFTR gene was recognized, cystic fibrosis was thought to be a disease of childhood.1 The diagnosis is now being made in adolescents and adults who have different clinical presentations than those observed in infants and young children. Many of these older patients have single-organ manifestations, such as pancreatitis, and do not fulfill the criteria for cystic fibrosis disease.
The penetrance of CFTR mutations is influenced by both genetic and environmental factors, and the clinical impact is organ-specific. The exocrine pancreas is the most consistent, though. Most patients with pancreatic manifestations of cystic fibrosis have severe mutations on both alleles. In persons with pancreatitis, the rationale for testing is to confirm or exclude cystic fibrosis and the traditional sweat test remains the primary diagnostic test. The identification of a CFTR mutation as the first-line test is likely to produce more questions than answers. For this reason, routine testing for CFTR mutations in patients with recurrent idiopathic pancreatitis is not recommended, but referral should be made to a cystic fibrosis center if this diagnosis is suspected.
Mutations in trypsinogen genes have also been associated with pancreatitis and are autosomal dominant.1 The cationic trypsinogen gene PRSS1 was discovered in 1996, and more than 20 mutations have been identified since that time. The more common mutations have a high penetrance, especially R112H and N29I. However, there is incomplete penetrance among many severe mutations and disease expression depends on other unknown factors. Because affected patients are at increased risk for pancreatic cancer, making the diagnosis provides some benefit. Testing is generally accepted for diagnosis in symptomatic persons, but screening of family members remains controversial. Currently, no guidelines exist for pancreatic cancer screening in these patients, but future developments could make the detection of this genetic abnormality more beneficial.
Chronic pancreatitis has also been associated with pancreatic secretory serine protease inhibitor Kazal type 1 (SPINK1), and more than 30 mutations have been identified.1 Most of these have low penetrance and are thought to cause disease predisposition rather than a causal relationship. There is no current indication for SPINK1 testing for diagnosis or screening.
 CELIAC SPRUE
CELIAC SPRUE
Celiac sprue affects 1 out of every 200 to 300 persons in Europe and North America.2 It is caused by an inappropriate T-cell mediated immune response against gluten in genetically predisposed persons. Malabsorption occurs when ingestion of wheat, rye, or barley gluten causes small intestine mucosal inflammation. The diagnosis is increasingly being made in adults, with 20% of new diagnoses in those over 60 years of age.
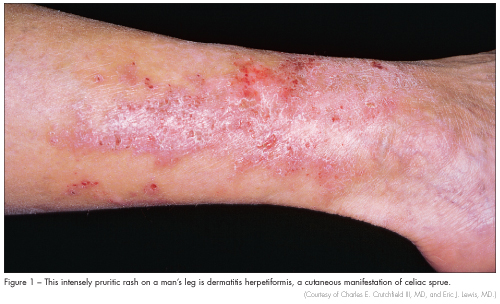
Celiac sprue should be suspected in patients with diarrhea, flatulence, weight loss, bloating, and abdominal discomfort. Many patients do not have gastrointestinal symptoms and present with asymptomatic iron deficiency anemia. Other laboratory abnormalities include folate deficiency, coagulopathy from vitamin K deficiency, and vitamin D deficiency. They may also present with dermatitis herpetiformis (Figure 1), characterized by pruritic papulovesicular lesions over the extensor surfaces of the extremities, buttocks, trunk, neck, and scalp. Because celiac sprue can become complicated by enteropathy-associated T-cell lymphoma and ulcerative jejunoileitis, accurate diagnosis and treatment is important.
Diagnosis can be made in different ways.2 Biopsy of normal-appearing skin in patients with dermatitis herpetiformis reveals granular deposits of IgA with immunofluorescence studies. Four serologic tests are available for diagnosis: IgA endomysial antibodies, IgA tissue transglutaminase (tTG) antibodies, IgA anti-gliadin antibodies (AGAs), and IgG AGAs.3 These can be used for diagnosis, screening, and evaluation of adherence. Small bowel biopsies remain the gold standard. Mucosal biopsies in celiac sprue are characterized by elongated intestinal crypts, flat mucosa, increased intraepithelial lymphocytes, and increased lymphocytes and plasma cells in the lamina propria (Figure 2).
 Celiac sprue is an HLA-associated disorder, but non-HLA genes are also thought to play a role.4 HLA-DQ is the main gene associated with celiac disease. Most carry a variant of DQ2, but some carry DQ8. These molecules bind and present peptide fragments to CD4+ T-helper cells. T cells in the small intestine of patients with celiac sprue uniquely recognize gluten peptides presented by DQ2 and DQ8, suggesting these HLA molecules preferentially present gluten peptides to CD4 T cells. The cells then produce many cytokines, including interferon-gamma, stimulating an array of inflammatory reactions that cause the lesions typical of celiac disease. In addition to adaptive immunity, innate immunity is stimulated in patients with celiac sprue. It is unclear which part of gluten stimulates this, but there is increased IL-15 in the lamina propria and epithelium in active disease.
Celiac sprue is an HLA-associated disorder, but non-HLA genes are also thought to play a role.4 HLA-DQ is the main gene associated with celiac disease. Most carry a variant of DQ2, but some carry DQ8. These molecules bind and present peptide fragments to CD4+ T-helper cells. T cells in the small intestine of patients with celiac sprue uniquely recognize gluten peptides presented by DQ2 and DQ8, suggesting these HLA molecules preferentially present gluten peptides to CD4 T cells. The cells then produce many cytokines, including interferon-gamma, stimulating an array of inflammatory reactions that cause the lesions typical of celiac disease. In addition to adaptive immunity, innate immunity is stimulated in patients with celiac sprue. It is unclear which part of gluten stimulates this, but there is increased IL-15 in the lamina propria and epithelium in active disease.
HLA genotype tests can be helpful when celiac sprue is suspected. The specificity and positive predictive value of these tests are poor. It is useless to predict celiac disease because many unaffected persons express the DQ abnormalities. However, HLA testing has a high negative predictive value and is useful in excluding the disease.
The treatment is a lifetime gluten-free diet.2 Approximately 70% of patients will have a clinical improvement within 2 weeks of starting the diet. Those with malabsorption may also need appropriate supplementation of vitamins A, D, E, K, iron, zinc, magnesium, and calcium. If patients do not respond to treatment, other diseases such as enteropathy-associated T-cell lymphoma should be considered.
PEUTZ-JEGHERS SYNDROME
Peutz-Jeghers syndrome is an autosomal dominant inherited disorder that is present in 1:8300 to 1:200,000 births.5 The disorder is characterized by gastrointestinal hamartomas and mucocutaneous pigmentation (Figure 3); affected patients are at increased risk for both gastrointestinal and extraintestinal malignancy.6 There is a wide variety of phenotypic expression in patients with this disease.
Peutz-Jeghers syndrome is caused by a germline mutation in the serine threonine kinase 11 gene (STK11), which can be found in 80% of clinically affected families.5 The gene is located on chromosome 19p13.3 and is a tumor suppressor gene. Genetic testing for clinical practice is widely available. There are currently no screening guidelines in place, but future developments could make identification of this genetic disease important for cancer prevention.
NEUROENDOCRINE TUMORS
Neuroendocrine tumors arise from the neuroendocrine cells of the endocrine system and comprise 2% of all malignant gastroenteropancreatic tumors.7 They are slow growing, but have malignant potential and are commonly not diagnosed until distant metastases are identified. Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) include carcinoid tumors and islet cell tumors. Many are sporadic, but some occur as part of genetic syndromes.
Multiple endocrine neoplasia-1 (MEN-1) syndrome is autosomal dominant and has high penetrance.7 The syndrome includes primary hyperparathyroidism, neuroendocrine pancreatic tumors (NPTs), and pituitary tumors. Ten percent of NPTs occur in association with MEN-1. Nonfunctioning NPTs and gastrinomas are strongly associated, but insulinomas, VIPomas, glucagonomas, and intestinal carcinoids are rarely associated with the disorder. MEN-1 results from an inactivation mutation in the MEN-1 gene, located on chromosome 11q13. It is a tumor suppressor gene and is caused by a germline mutation of the gene followed by somatic inactivation of the unaffected allele. Once the diagnosis is established in a patient, a MEN-1 germline mutation DNA test should be performed in all family members after the first decade of life. Those identified with the mutation should be screened regularly with annual biochemical testing and imaging every 3 years.
Von Hippel-Lindau syndrome is autosomal dominant and is characterized by retinal or CNS hemangioblastomas, clear cell renal carcinomas, pheochromocytomas, and pancreatic cystic tumors or NPTs.7 Pancreatic lesions are seen in 20% to 75% of affected patients. The von Hippel-Lindau gene is a tumor suppressor gene and is located on chromosome 3p25-26. It regulates hypoxia-induced cell proliferation and angiogenesis. Once the diagnosis is established, DNA testing of family members is required. These patients should also undergo regular clinical screening even if asymptomatic with regular ophthalmoscopic examinations, 24-hour urine catecholamine measurement, and brain and abdominal imaging.
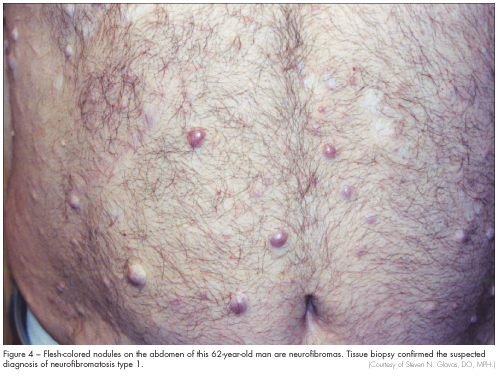 Neurofibromatosis (NF-1) is autosomal dominant and is characterized by café au lait spots, cutaneous or subcutaneous neurofibromas (Figure 4), optic gliomas, benign iris hamartomas, and dysplastic bone lesions.7 Ampullary carcinoids, duodenal and pancreatic somatostatinomas, pheochromocytomas, and paragangliomas may also develop. The NF-1 gene is a tumor suppressor gene and is located on chromosome 17q11.2. It encodes a protein called neurofibromin. Penetrance of NF-1 is variable, but screening seems to be worthwhile in affected families.
Neurofibromatosis (NF-1) is autosomal dominant and is characterized by café au lait spots, cutaneous or subcutaneous neurofibromas (Figure 4), optic gliomas, benign iris hamartomas, and dysplastic bone lesions.7 Ampullary carcinoids, duodenal and pancreatic somatostatinomas, pheochromocytomas, and paragangliomas may also develop. The NF-1 gene is a tumor suppressor gene and is located on chromosome 17q11.2. It encodes a protein called neurofibromin. Penetrance of NF-1 is variable, but screening seems to be worthwhile in affected families.
Tuberous sclerosis is autosomal dominant and is characterized by hamartomas, astrocytomas, and well-differentiated tumors of the brain, heart, skin, kidney, lung, and pancreas.7 Affected patients usually present with epilepsy and mental retardation in childhood. The associated genes are TSC1 and TSC2. TSC1 is located on chromosome 9q34 and encodes hamartin. TSC2 is located on chromosome 16p13.3 and encodes tuberin. DNA testing is available; however, there is a 15% false-negative rate and germline mosaicism in about 2% of affected persons.
HEREDITARY HEMORRHAGIC TELANGIECTASIA
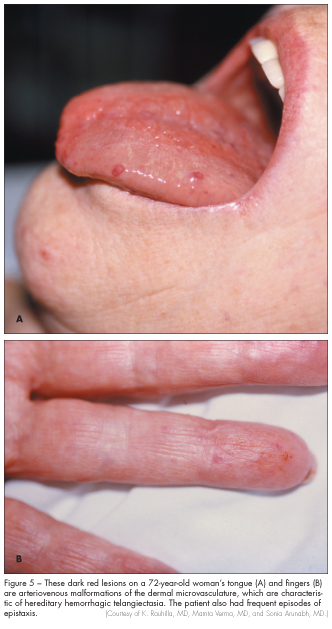
Hereditary hemorrhagic telangiectasia, also known as Rendu-Osler-Weber syndrome, was once thought to be rare.8 However, it is now believed to be more common. The manifestations of hereditary hemorrhagic telangiectasia result from abnormalities of vascular structure in the nose, skin, lung, brain, and gastrointestinal tract. The lesions range from small, focal dilatations of postcapillary venules to markedly dilated and convoluted venules. Common manifestations of this disorder include epistaxis and telangiectasias of the lips, tongue, fingers, face, nail beds, arms, or face (Figure 5). Pulmonary arteriovenous malformations occur in 5% to 15% of patients with hereditary hemorrhagic telangiectasia, resulting in direct right-to-left shunts. The symptoms include dyspnea, fatigue, cyanosis, and polycythemia. Neurologic symptoms, such as migraines, transient ischemic attacks, strokes, and intracerebral or subarachnoid hemorrhage, can occur as a result of pulmonary abnormalities or from vascular abnormalities in the brain itself.
 Gastrointestinal manifestations are less common, but they are some of the most difficult to manage. Patients usually present with recurrent hemorrhage of the upper or lower gastrointestinal tract in the fifth or sixth decade of life. Endoscopy may reveal telangiectasias in the stomach, small bowel, or colon that are similar to those in the mouth or nose. The liver may also be involved, which can lead to atypical cirrhosis.
Gastrointestinal manifestations are less common, but they are some of the most difficult to manage. Patients usually present with recurrent hemorrhage of the upper or lower gastrointestinal tract in the fifth or sixth decade of life. Endoscopy may reveal telangiectasias in the stomach, small bowel, or colon that are similar to those in the mouth or nose. The liver may also be involved, which can lead to atypical cirrhosis.
Hereditary hemorrhagic telangiectasia has been linked to chromosome 9q33-34 in some families and chromosome 12q in others, but other genes may be involved.8 The gene on 9q3 is identified as endoglin and encodes an integral membrane glycoprotein that is found in endothelial cells to bind transforming growth factor beta. Transforming growth factor beta functions in migration, proliferation, and adhesion of endothelial cells as well as in composition and organization of the extracellular matrix.
Hereditary hemorrhagic telangiectasia is autosomal dominant but with incomplete penetrance. At this time, diagnosis is still clinical. Patients must have two of the following: recurrent epistaxis, telangiectasias somewhere besides the nasal mucosa, evidence of autosomal dominance in the family, and visceral involvement.
INFLAMMATORY BOWEL DISEASE
Crohn’s disease and ulcerative colitis are characterized by both intestinal and extra-intestinal manifestations. These patients have inflammation of the gastrointestinal tract because of a dysregulated mucosal immune response.9 There are racial and ethnic differences in incidence, with an increased risk in Western countries, urban areas, and higher socioeconomic classes. In the United States, Caucasians are at higher risk than other races. There is also clustering within families; 5% to 10% of those with inflammatory bowel disease have a family history. A positive family history is the greatest risk factor, which suggests a genetic component.
Inflammatory bowel disease is a complex genetic disorder, and multiple genes influence disease expression. Crohn’s disease has been associated with the inflammatory bowel disease locus in the pericentromeric region of chromosome 16, which contains the nucleotide-binding oligomerization domain-containing protein 2/caspase recruitment domain-containing protein 15 gene. It was first identified in 1996 and its association with Crohn’s disease has been replicated in numerous studies. The inflammatory bowel disease 2 locus on chromosome 12 has been linked more to ulcerative colitis than to Crohn’s disease, but replication has not been universal. The inflammatory bowel disease 3 locus, found on chromosome 6p, encompasses the major histocompatibility complex and has been implicated in both ulcerative colitis and Crohn’s disease in a number of studies.10 This region contains the tumor necrosis factor gene and has been observed more frequently in affected males than females. The inflammatory bowel disease 5 locus is in the chromosome 5q31-q33 region, but the causative genes have not been established. It has been associated with early-onset Crohn’s disease within Caucasian families in Canada. The incidence of Crohn’s disease is increased 2-fold in heterozygotes and 6-fold in homozygotes. This region contains several immunoregulatory cytokines that might be important in the development of Crohn’s disease. There are other loci that have been identified. Chromosomes 19p13, 1p, 7q, and 3p have been linked to inflammatory bowel disease, but further investigation is needed.
The evidence is greatest for association of Crohn’s disease with the NOD2/CARD15 gene. It is expressed in peripheral blood monocytes. The N-terminus portion contains 2 CARD domains that mediate protein-protein interactions: the central nucleotide-binding domain mediates self-oligomerization for activation, and the C-terminus leucine-rich repeat domain recognizes microbial components. There are three coding polymorphisms within the C-terminus that have been associated with Crohn’s disease: Arg702Trp, Gly908Arg, and Leu1007fsinsC. One copy of these alleles is associated with a 2- to 4-fold increased risk of Crohn’s disease. Having two copies increases the risk 20- to 40-fold, indicating an autosomal recessive inheritance. Approximately 8% to 17% of patients with Crohn’s disease have two copies. These alleles are associated with both familial cases of Crohn’s disease and sporadic cases.
 The chromosome 6p region containing the major histocompatibility complex has been intensely studied. This HLA region is highly polymorphic and exhibits genetic differences between individuals. Because of the enormous immunologic complexity in this region, identification of specific disease associations has been difficult. There is evidence that specific HLA II associations contribute to inflammatory bowel disease, especially ulcerative colitis. Ulcerative colitis has been associated with DR9 and DRB1*0103. Crohn’s disease has been associated with DR7, DRB3*0301, and DQ4. It has also been suggested that HLA genes are associated with disease location in ulcerative colitis and extraintestinal manifestations.11
The chromosome 6p region containing the major histocompatibility complex has been intensely studied. This HLA region is highly polymorphic and exhibits genetic differences between individuals. Because of the enormous immunologic complexity in this region, identification of specific disease associations has been difficult. There is evidence that specific HLA II associations contribute to inflammatory bowel disease, especially ulcerative colitis. Ulcerative colitis has been associated with DR9 and DRB1*0103. Crohn’s disease has been associated with DR7, DRB3*0301, and DQ4. It has also been suggested that HLA genes are associated with disease location in ulcerative colitis and extraintestinal manifestations.11
Epidemiologic data suggest that genetic factors are a major component of the development of inflammatory bowel disease. Important advances have been made in identifying some of the contributing genes, but the mechanisms are complex and there is still progress to be made. Screening of unaffected relatives is not recommended at this time because of the limited penetrance of the NOD2 genotypes.
GENETIC TESTING AND COUNSELING
Medicine has entered a new era, with many genetic tests available to assist clinicians. Many tests are already commercially available (Table), but many more are sure to follow. Physicians can now obtain detailed genetic information on patients, which is leading to personalized medicine. This information can be used for prenatal diagnosis, newborn screening, carrier screening, diagnosis, and pharmacogenetics.12 As information about these tests becomes increasingly available, patients will surely have many questions regarding these tests and how they can be of benefit.
Genetic counseling should be offered with genetic testing. There are psychological effects in knowing that one is genetically predisposed to certain cancers and other life-altering conditions. This may lead to depression and changes in quality of life and can cause patients to avoid having children. This may also affect patients’ ability to obtain health or life insurance, although they should be legally protected. There are times when this information may be more harmful than helpful; thus, the decision to perform genetic testing should be handled on an individual basis.
1. Ooi CY, Gonska T, Durie PR, Freedman SD. Genetic testing in pancreatitis. Gastroenterology. 2010;138:2202-2206.
2. Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002;346(3):180-188.
3. Farrell RJ, Kelly CP. Diagnosis of celiac sprue. Am J Gastroenterol. 2001;96(12):3237-3246.
4. Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol. 2005;3:843-851.
5. van Lier MGF, Wagner A, Mathus-Vliegen EMH, et al. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol.2010;105:1258-1264.
6. Boardman LA, Couch FJ, Burgart LJ, et al. Genetic heterogeneity in Peutz-Jeghers syndrome. Human Mutation. 2000;16:23-30.
7. Toumpanakis CG, Caplin ME. Molecular genetics of gastroenteropancreatic neuroendocrine tumors.Am J Gatroenterol. 2008;103:729-732.
8. Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med.1995;333(14):918-924.
9. Bonen DK, Cho JH. The genetics of inflammatory bowel disease. Gastroenterology.2003;124:521-536.
10. Ahmad T, Satsangi J, McGovern D, Bunce M, Jewell DP. Review article: the genetics of inflammatory bowel disease. Aliment Pharmacol Ther. 2001;15:731-748.
11. van Limbergen J, Philoptt D, Griffiths AM. Genetic profiling in inflammatory bowel disease: from association to bedside. Gastroenterology.2001;141:1566-1571.
12. Lerman C, Croyle RT, Tercyak KP, Hamman H. Genetic testing: psychological aspects and implications. J Consulting Clin Psychology. 2002;70(3):784-797.